INTRODUCTION TO MOLECULAR BIOLOGY
DATABASES AND SEQUENCE ANALYSIS
9-13 March 1998
Tore Samuelsson
Göteborg University
Dept of Medical Biochemistry
The flow of genetic information and bioinformatics
DNA -> RNA -> protein -> conformation
Databases in bioinformatics
- Structure
PDB (Protein Data Bank, Brookhaven Natl Lab,
www.pdb.bnl.gov)
Xray
crystallography
NMR
modeling
KLOTHO
(small molecules, www.ibc.wustl.edu/klotho/)
- Sequence
DNA
Genbank
(www.ncbi.nlm.nih.gov)
- Homologisökning:
/BLAST/)
- Entrez : /Entrez/
GSDB (Genome sequence database)
EMBL (European Molecular Biology
Laboratory, www.ebi.ac.uk)
-
SRS : srs.ebi.ac.uk:5000
DDBJ
(DNA Data Bank of Japan)
Protein
Swissprot
(www.ebi.ac.uk)
- Genome
GDB
(Human Genome Data Base, gdbwww.gdb.org)
Mouse
genome database (www.informatics.jax.org)
Yeast
genome (genome-ftp.stanford.edu/Saccharomyces)
Bacterial
genomes (www.tigr.org)
- Genetic disorders
OMIM
(Online Mendelian Inheritance in Man,
www.ncbi.nlm.nih.gov)
- Taxonomy
(www.ncbi.nlm.nih.gov)
- Prosite (expasy.hcuge.ch)
- Literature
Medline
(www.ncbi.nlm.nih.gov)
Structure databases.
The Brookhaven Protein Data Bank
PDB: Number of Entries
Deposited and Released by Year
as of 02/11/98
|
Year
|
Deposited
|
Released
|
|
1973
|
10
|
10
|
|
1974
|
7
|
2
|
|
1975
|
18
|
21
|
|
1976
|
47
|
27
|
|
1977
|
27
|
38
|
|
1978
|
26
|
26
|
|
1979
|
32
|
31
|
|
1980
|
20
|
30
|
|
1981
|
39
|
26
|
|
1982
|
59
|
47
|
|
1983
|
27
|
50
|
|
1984
|
36
|
29
|
|
1985
|
27
|
30
|
|
1986
|
28
|
29
|
|
1987
|
64
|
28
|
|
1988
|
129
|
79
|
|
1989
|
192
|
89
|
|
1990
|
306
|
164
|
|
1991
|
512
|
205
|
|
1992
|
635
|
226
|
|
1993
|
922
|
849
|
|
1994
|
1111
|
1392
|
|
1995
|
1221
|
1002
|
|
1996
|
1448
|
1224
|
|
1997
|
1848
|
1640
|
|
1998
|
207
|
262
|
NOTE: Computing the totals for number of entries released will produce a sum
greater than what is currently available from PDB. This is due to entries being
withdrawn and/or replaced.
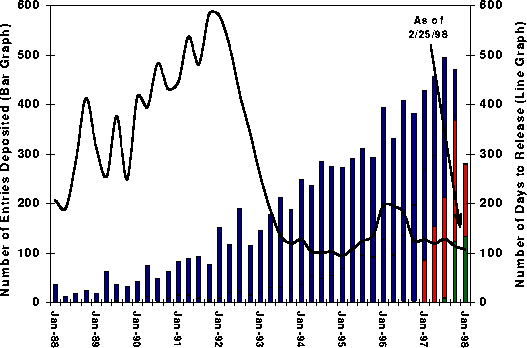
Number of Entries Deposited (Bar)
and Average Time to Release (Line)
Accumulated and Averaged on a Quarterly Basis
Bar Graph -
Number of Entries in the Following Categories:
·
OnHold - (red) On-hold per depositor request
·
Processing - (green) Being
processed
·
Released - (blue) Released
Line
Graph
AVERAGE - Average number of days to release
The data were
accumulated and averaged on a quarterly basis. The average turn around times
for entries now being processed are estimated based on the average of the last
12 months.
Data for the
last quarter are accumulated until the date specified on the graph.
View Data in Tabular
Form Statistics
on contents and growth of the PDB

PDB Holdings List
Entries loaded on
February 25, 1998
7163
Released Atomic Coordinate Entries
1736
Structure Factor Files
400 NMR Restraint Files
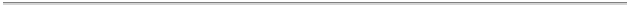
Molecule
Type
6339 proteins, peptides, and viruses
284 protein/nucleic acid complexes
528 nucleic acids
12 carbohydrates
0 others
Experimental
Technique
5850 diffraction and other
1133 NMR
180 theoretical modeling
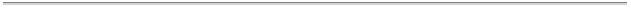
Count
By Experiment
Diffraction
5261 proteins,peptides, and viruses
346 nucleic acids
233 protein/nucleic acid complexes
10 carbohydrates
0 others
NMR
922 proteins,peptides, and viruses
167 nucleic acids
42 protein/nucleic acid complexes
2 carbohydrates
0 others
Model
156 proteins,peptides, and viruses
15 nucleic acids
9 protein/nucleic acid complexes
0 carbohydrates
0 others
Example of PDB entry
HEADER
HORMONE
30-OCT-92 1BPH 1BPH
2
COMPND INSULIN
(CUBIC) IN 0.1M SODIUM SALT SOLUTION AT PH9 1BPH 3
SOURCE BOVINE
(BOS $TAURUS) PANCREAS 1BPH 4
AUTHOR
O.GURSKY,J.BADGER,Y.LI,D.L.D.CASPAR 1BPH
5
REVDAT 2 31-OCT-93 1BPHA 1 REMARK HET FORMUL 1BPHA 1
REVDAT 1 15-JAN-93 1BPH 0 1BPH 6
JRNL
AUTH
O.GURSKY,J.BADGER,Y.LI,D.L.D.CASPAR 1BPH 7
JRNL
TITL CONFORMATIONAL CHANGES IN
CUBIC INSULIN CRYSTALS 1BPH 8
JRNL TITL 2
IN THE PH RANGE 7-11 1BPH 9
JRNL
REF BIOPHYS.J.
V. 63 1210 1992 1BPH 10
JRNL
REFN ASTM BIOJAU US ISSN 0006-3495 030
1BPH 11
REMARK 1
1BPH 12
REMARK 1
REFERENCE 1 1BPH 13
REMARK 1 AUTH
O.GURSKY,Y.LI,J.BADGER,D.L.D.CASPAR 1BPH 14
REMARK 1 TITL
MONOVALENT CATION BINDING IN CUBIC INSULIN 1BPH 15
REMARK 1 TITL 2 CRYSTALS 1BPH 16
REMARK 1 REF
BIOPHYS.J.
V. 61 604 1992 1BPH 17
REMARK 1 REFN
ASTM BIOJAU US ISSN
0006-3495 030 1BPH
18
REMARK 1
REFERENCE 2 1BPH 19
REMARK 1 AUTH
J.BADGER 1BPH 20
REMARK 1 TITL
FLEXIBILITY IN CRYSTALLINE INSULINS 1BPH 21
REMARK 1 REF
BIOPHYS.J. V.
61 816 1992 1BPH
22
REMARK 1 REFN
ASTM BIOJAU US ISSN
0006-3495 030 1BPH
23
REMARK 1
REFERENCE 3 1BPHA 2
REMARK 1 AUTH
J.BADGER,M.R.HARRIS,C.D.REYNOLDS,A.C.EVANS, 1BPH 25
REMARK 1 AUTH 2
E.J.DODSON,G.G.DODSON,A.C.T.NORTH 1BPH 26
REMARK 1 TITL
STRUCTURE OF THE PIG INSULIN DIMER IN THE CUBIC 1BPH
27
REMARK 1 TITL 2 CRYSTAL 1BPH 28
REMARK 1 REF
ACTA CRYSTALLOGR.,SECT.B
V. 47 127 1991 1BPH 29
REMARK 1 REFN
ASTM ASBSDK DK ISSN
0108-7681 622 1BPH
30
REMARK 1
REFERENCE 4 1BPHA 3
REMARK 1 AUTH
J.BADGER,D.L.D.CASPAR 1BPH 32
REMARK 1 TITL
WATER STRUCTURE IN CUBIC INSULIN CRYSTALS 1BPH 33
REMARK 1 REF
PROC.NAT.ACAD.SCI.USA V.
88 622 1991 1BPH 34
REMARK 1 REFN
ASTM PNASA6 US ISSN
0027-8424 040 1BPH
35
REMARK 1
REFERENCE 5 1BPHA 4
REMARK 1 AUTH
E.J.DODSON,G.G.DODSON,A.LEWITOVA,M.SABESAN 1BPH 37
REMARK 1 TITL
ZINC-FREE CUBIC PIG INSULIN: CRYSTALLIZATION AND 1BPH
38
REMARK 1 TITL 2 STRUCTURE DETERMINATION 1BPH 39
REMARK 1 REF
J.MOL.BIOL. V.
125 387 1978 1BPH 40
REMARK 1 REFN
ASTM JMOBAK UK ISSN
0022-2836 070 1BPH
41
REMARK 2
1BPH 42
REMARK 2
RESOLUTION. 2.0 ANGSTROMS. 1BPH 43
REMARK 3 1BPH 44
REMARK 3
REFINEMENT. 1BPH 45
REMARK 3 PROGRAM PROLSQ 1BPH
46
REMARK 3 AUTHORS HENDRICKSON AND KONNERT 1BPH 47
REMARK 3 R VALUE 0.160 1BPH
48
REMARK 3 RMSD BOND DISTANCES 0.014
ANGSTROMS
1BPH 49
REMARK 3 RMSD BOND ANGLE DISTANCES 0.043
ANGSTROMS
1BPH 50
REMARK 4
1BPH 51
REMARK 4 THIS
CRYSTAL FORM CONTAINS ONE INSULIN MOLECULE PER 1BPH 52
REMARK 4
ASYMMETRIC UNIT. THE SOLVENT VOLUME IS
64 PERCENT OF THE 1BPH 53
REMARK 4 CRYSTAL
VOLUME. THERE ARE MANY ALTERED SIDE CHAIN TORSION 1BPH 54
REMARK 4 ANGLES
AND MAIN CHAIN DISPLACEMENTS IN THE CUBIC CRYSTAL 1BPH 55
REMARK 4
STRUCTURE COMPARED TO OTHER INSULIN CRYSTAL FORMS. ABOUT 1BPH 56
REMARK 4 30 PER
CENT OF THE AMINO ACID RESIDUES CAN ADOPT MULTIPLE 1BPH 57
REMARK 4
CONFORMATIONS WHICH WERE RELIABLY IDENTIFIED BY COMPARISON 1BPH
58
REMARK 4 OF THE
DATA SETS COLLECTED FROM THE CRYSTALS IN THE PH 1BPH 59
REMARK 4 RANGE 7
- 11. THE WEIGHTS OF MANY OF SUCH
MULTIPLE PROTEIN 1BPH 60
REMARK 4 AND
SOLVENT CONFORMATIONS DEPEND ON SOLVENT IONIC 1BPH 61
REMARK 4
CONDITIONS (PH AND SALT CONCENTRATION). 1BPH
62
REMARK 5
1BPH 63
REMARK 5 THERE
ARE FOUR RELATED ENTRIES: 1BPH
64
REMARK 5 1APH
0.1M SODIUM SALT SOLUTION AT PH 7 1BPH 65
REMARK 5 1BPH
0.1M SODIUM SALT SOLUTION AT PH 9 1BPH 66
REMARK 5 1CPH
0.1M SODIUM SALT SOLUTION AT PH 10 1BPH 67
REMARK 5 1DPH
1.0M SODIUM SALT SOLUTION AT PH 11 1BPH 68
REMARK 6 1BPH 69
REMARK 6 IN 1BPH
AND 1CPH, THE SIDE CHAIN OF GLU A 4 CAN ADOPT TWO 1BPH 70
REMARK 6
ALTERNATIVE POSITIONS WHICH OVERLAP.
THEIR RELATIVE WEIGHT 1BPH 71
REMARK 6 AND THE
ATOMIC POSITIONS OF THE SECOND CONFORMER ARE NOT 1BPH 72
REMARK 6
ACCURATELY DETERMINED. 1BPH 73
REMARK 7
1BPH 74
REMARK 7 IN
1APH, 1BPH, AND 1DPH, THE SIDE CHAIN OF GLU B 21 IS 1BPH 75
REMARK 7
DISORDERED. IT HAS BEEN MODELED AS
SUPERPOSITION OF TWO 1BPH 76
REMARK 7
CONFORMATIONS BUT ATOMIC POSITIONS FOR THESE CONFORMATIONS 1BPH
77
REMARK 7 ARE
PROBABLY NOT VERY ACCURATE. 1BPH
78
REMARK 8
1BPH 79
REMARK 8 THE
SIDE CHAIN OF LYS B 29 IS POORLY DEFINED IN THE 1BPH 80
REMARK 8
ELECTRON DENSITY MAPS. IN 1APH AND
1CPH, IT IS INCLUDED 1BPH 81
REMARK 8 WITH
PARTIAL OCCUPANCY. IN 1BPH AND 1DPH,
ITS COORDINATES 1BPH 82
REMARK 8 HAVE
BEEN OMITTED FROM THE ENTRY. 1BPH
83
REMARK 13 1BPHA 5
REMARK 13
CORRECTION. RENUMBER REFERENCES SEQUENTIALLY.
INSERT 1BPHA 6
REMARK 13 MISSING HET AND FORMUL RECORDS FOR NA. 31-OCT-93. 1BPHA 7
SEQRES 1 A 21
GLY ILE VAL GLU GLN CYS CYS ALA SER VAL CYS SER LEU 1BPH 106
SEQRES 2 A 21
TYR GLN LEU GLU ASN TYR CYS ASN 1BPH 107
SEQRES 1 B 30
PHE VAL ASN GLN HIS LEU CYS GLY SER HIS LEU VAL GLU 1BPH 108
SEQRES 2 B 30
ALA LEU TYR LEU VAL CYS GLY GLU ARG GLY PHE PHE TYR 1BPH 109
SEQRES 3 B 30
THR PRO LYS ALA 1BPH 110
HET DCE 200
4
1,2-DICHLOROETHANE(ETHYLENE DICHLORIDE)
1BPH 111
HET NA 88
1 SODIUM ION 1BPHA 8
FORMUL 3 DCE
C2 H4 CL2 1BPH 112
FORMUL 4 NA
NA1 1BPHA 9
FORMUL 5 HOH
*55(H2 O1) 1BPHA 10
HELIX 1 A1 GLY A
1 VAL A 10 1 1BPH 114
HELIX 2 A2 SER A
12 GLU A 17
5 NOT IDEAL
1BPH 115
HELIX 3 B1 SER B
9 GLY B 20
1
1BPH 116
TURN 1 1B1 CYS
B 19
ARG B 22 1BPH 117
TURN 2 1B2 GLY
B 20
GLY B 23 1BPH 118
SSBOND 1 CYS
A 6 CYS A 11 1BPH 119
SSBOND 2 CYS
A 7 CYS B 7 1BPH 120
SSBOND 3 CYS
A 20 CYS B 19 1BPH 121
CRYST1
78.900 78.900 78.900
90.00 90.00 90.00 I 21 3 24 1BPH 122
ORIGX1
1.000000 0.000000 0.000000 0.00000
1BPH 123
ORIGX2
0.000000 1.000000 0.000000 0.00000
1BPH 124
ORIGX3
0.000000 0.000000 1.000000 0.00000
1BPH 125
SCALE1
0.012674 0.000000 0.000000 0.00000
1BPH 126
SCALE2
0.000000 0.012674 0.000000 0.00000
1BPH 127
SCALE3
0.000000 0.000000 0.012674 0.00000
1BPH 128
ATOM 1 N
GLY A 1 13.994
47.196 31.798 1.00 35.87 1BPH 129
ATOM 2 CA
GLY A 1 14.277
46.226 30.708 1.00 38.67 1BPH 130
ATOM 3 C
GLY A 1 15.574
45.507 31.085 1.00 31.18 1BPH 131
ATOM 4 O
GLY A 1 16.078
45.660 32.217 1.00 22.60 1BPH 132
ATOM 5 N
ILE A 2 16.088
44.766 30.126 1.00 28.39 1BPH 133
ATOM 6 CA
ILE A 2 17.342
44.034 30.404 1.00 23.76 1BPH 134
ATOM 7 C
ILE A 2 18.526
44.939 30.686 1.00 25.29 1BPH 135
ATOM 8 O
ILE A 2 19.425
44.457 31.392 1.00 18.74 1BPH 136
ATOM 9 CB
ILE A 2 17.571
43.072 29.158 1.00 27.36 1BPH 137
ATOM 10 CG1 ILE A
2 18.638 42.049
29.605 1.00 18.03 1BPH 138
ATOM 11 CG2 ILE A
2 17.859 43.936
27.903 1.00 25.54 1BPH 139
ATOM 12 CD1 ILE A
2 18.914 40.930
28.590 1.00 17.07 1BPH 140
ATOM 13 N
VAL A 3 18.619
46.195 30.192 1.00 24.42 1BPH 141
ATOM 14 CA
VAL A 3 19.774
47.080 30.436 1.00 30.26 1BPH 142
ATOM 15 C
VAL A 3 19.952
47.453 31.895 1.00 19.08 1BPH 143
ATOM 16 O
VAL A 3 21.018
47.421 32.561 1.00 28.15 1BPH 144
ATOM 17 CB
VAL A 3 19.719
48.274 29.462 1.00 33.87 1BPH 145
ATOM 18 CG1 VAL A
3 20.847 49.225
29.754 1.00 30.40 1BPH 146
ATOM 19 CG2 VAL A
3 19.868 47.724
28.044 1.00 24.51 1BPH 147
.
.
.
ATOM 127
N GLU A 17
17.257 34.367 30.913
1.00 17.57 1BPH 255
ATOM 128 CA
GLU A 17 16.353
33.393 30.338 1.00 13.26 1BPH 256
ATOM 129 C
GLU A 17 14.968
33.889 30.001 1.00 22.70 1BPH 257
ATOM 130 O
GLU A 17 14.234
33.275 29.212 1.00 25.00 1BPH 258
ATOM 131 CB
GLU A 17 16.183
32.146 31.209 1.00 17.01 1BPH 259
ATOM 132 CG
GLU A 17 17.252
31.160 30.695 1.00 14.38 1BPH 260
ATOM 133 CD
GLU A 17 16.968
29.843 31.385 1.00 24.91 1BPH 261
ATOM 134 OE1 GLU A
17 16.230 29.713
32.350 1.00 25.72 1BPH 262
ATOM 135 OE2 GLU A
17 17.675 28.984
30.830 1.00 22.42 1BPH 263
ATOM 136 N
ASN A 18 14.618
35.021 30.563 1.00 22.30 1BPH 264
ATOM 137 CA
ASN A 18 13.371
35.753 30.369 1.00 29.65 1BPH 265
ATOM 138 C
ASN A 18 13.330
36.318 28.943 1.00 23.17 1BPH 266
ATOM 139 O
ASN A 18 12.197
36.611 28.486 1.00 30.58 1BPH 267
ATOM 172 N
PHE B 1 28.961
32.694 34.302 1.00 38.09 1BPH 300
ATOM 173 CA
PHE B 1 29.545
33.933 33.691 1.00 44.75 1BPH 301
ATOM 174 C
PHE B 1 28.483
35.030 33.562 1.00 18.46 1BPH 302
ATOM 175 O
PHE B 1 28.656
36.170 33.083 1.00 29.15 1BPH 303
ATOM 176 CB
PHE B 1 30.190
33.486 32.346 1.00 36.50 1BPH 304
ATOM 177 CG
PHE B 1 29.191
32.986 31.322 1.00 29.77 1BPH 305
ATOM 178 CD1 PHE B
1 28.691 31.688
31.351 1.00 22.29 1BPH 306
ATOM 179 CD2 PHE B
1 28.736 33.844
30.327 1.00 30.11 1BPH 307
ATOM 180 CE1 PHE B
1 27.758 31.234
30.415 1.00 30.11 1BPH 308
ATOM 181 CE2 PHE B
1 27.822 33.423
29.377 1.00 29.49 1BPH 309
ATOM 182 CZ
PHE B 1 27.329
32.125 29.428 1.00 27.29 1BPH 310
ATOM 183 N
VAL B 2 27.235
34.671 33.935 1.00 25.09 1BPH 311
ATOM 184 CA
VAL B 2 26.085
35.571 33.793 1.00 23.88 1BPH 312
ATOM 185 C
VAL B 2 25.902
36.506 34.969 1.00 24.42 1BPH 313
ATOM 186 O
VAL B 2 25.269
37.560 34.801 1.00 19.63 1BPH 314
ATOM 187 CB
VAL B 2 24.846
34.751 33.391 1.00 28.89 1BPH 315
.
ATOM 413 N
PRO B 28 16.809
47.082 24.129 1.00 39.30 1BPH 541
ATOM 414 CA
PRO B 28 17.550
47.958 25.065 1.00 50.32 1BPH 542
ATOM 415 C
PRO B 28 16.747
49.100 25.692 1.00 51.41 1BPH 543
ATOM 416 O
PRO B 28 16.922
49.526 26.848 1.00 52.87 1BPH 544
ATOM 417 CB
PRO B 28 18.744
48.435 24.231 1.00 33.07 1BPH 545
ATOM 418 CG
PRO B 28 18.261
48.353 22.779 1.00 28.91 1BPH 546
ATOM 419 CD
PRO B 28 17.355
47.133 22.751 1.00 30.72 1BPH 547
ATOM 420 N
LYS B 29 15.830
49.593 24.905 1.00 58.03 1BPH 548
ATOM 421 CA ALYS B
29 14.935 50.708
25.214 0.50 56.38 1BPH 549
ATOM 422 CA BLYS B
29 15.106 50.841
24.970 0.50 57.81 1BPH 550
ATOM 423 C
ALYS B 29 13.602
50.396 25.876 0.50 73.09 1BPH 551
ATOM 424 C
BLYS B 29 13.915
50.201 25.692 0.50 66.40 1BPH 552
ATOM 425 O
ALYS B 29 13.044
51.332 26.517 0.50 80.92 1BPH 553
ATOM 426 O
BLYS B 29 12.908
49.842 25.053 0.50 53.34 1BPH 554
ATOM 427 CB ALYS B
29 14.689 51.541
23.932 0.50 58.98 1BPH 555
ATOM 428 CB BLYS B
29 14.658 51.386
23.598 0.50 45.66 1BPH 556
ATOM 429 N
AALA B 30 13.056
49.194 25.782 0.50 74.55 1BPH 557
ATOM 430 N
BALA B 30 14.075
50.102 27.005 0.50 71.75 1BPH 558
ATOM 431 CA AALA B
30 11.762 48.878
26.416 0.50 75.29 1BPH 559
ATOM 432 CA BALA B
30 13.075 49.536
27.915 0.50 73.80 1BPH 560
ATOM 433 C
AALA B 30 11.853
47.818 27.515 0.50 68.10 1BPH 561
ATOM 434 C
BALA B 30 12.867
50.426 29.144 0.50 73.94 1BPH 562
ATOM 435 O
AALA B 30 10.774
47.235 27.799 0.50 65.90 1BPH 563
ATOM 436 O
BALA B 30 12.394
49.828 30.144 0.50 69.68 1BPH 564
ATOM 437 CB AALA B
30 10.728 48.457
25.375 0.50 76.93 1BPH 565
ATOM 438 CB BALA B
30 13.512 48.144
28.366 0.50 73.70 1BPH 566
ATOM 439 OXTAALA B
30 12.952 47.610
28.048 0.50 63.45 1BPH 567
ATOM 440 OXTBALA B
30 13.182 51.623
29.061 0.50 76.41 1BPH 568
TER 441 ALA B
30 1BPH 569
HETATM 442
CL1 DCE 200 26.950 41.213
19.536 0.50 34.85 1BPH 570
HETATM 443 C1
DCE 200 28.222 40.003 20.178 0.50 24.42 1BPH 571
HETATM 444 C2
DCE 200 28.307
38.776 19.363 0.50 24.99 1BPH 572
HETATM 445
CL2 DCE 200 26.941 37.681
19.833 0.50 33.75 1BPH 573
HETATM 446
NA NA 88 20.339 43.145
38.263 0.50 13.22 1BPH 574
HETATM 447 O
HOH 1 26.102
28.408 28.110 0.33 28.57 1BPH 575
HETATM 448 O
HOH 2 26.719
28.525 28.242 0.66 30.29 1BPH 576
HETATM 449 O
HOH 3 19.213
33.037 38.295 1.00 42.10 1BPH 577
HETATM 450 O
HOH 4 21.104
32.216 20.645 1.00 26.61 1BPH 578
HETATM 451 O
HOH 5 21.954
33.637 38.117 1.00 22.77 1BPH 579
HETATM 498 O
HOH 52 19.217
52.503 35.050 1.00 68.12 1BPH 626
HETATM 499 O
HOH 53 15.376
24.434 25.540 1.00 82.81 1BPH 627
HETATM 500 O
HOH 54 21.768
55.234 32.076 1.00 85.97 1BPH 628
HETATM 501 O
HOH 55 22.667
52.737 33.359 1.00 81.22 1BPH 629
CONECT 48 47
78 1BPH 630
CONECT 54 53
235 1BPH 631
CONECT 78 48
77 1BPH 632
CONECT 161 160
331 1BPH 633
CONECT 235 54
234 1BPH 634
CONECT 331 161
330 1BPH 635
CONECT 442 443 1BPH 636
CONECT 443 442
444 1BPH 637
CONECT 444 443
445 1BPH 638
CONECT 445 444 1BPH 639
MASTER
97 0 2 3 0
2 0 6 499 2
10 5 1BPHA 11
END
1BPH
3D viewers
Rasmol www.umass.edu/microbio/rasmol/
Weblab www.msi.com
Kinemage www.cryst.bbk.ac.uk/PPS/vsns-pps/technology/kinemage.html
Chime www.umass.edu/microbio/rasmol/
----
Sequence databases
- DNA Sequence
Genbank
(www.ncbi.nlm.nih.gov)
GSDB
(Genome sequence database)
EMBL (European Molecular Biology
Laboratory)(www.ebi.ac.uk)
DDBJ
(DNA Data Bank of Japan)
Current statistics of EMBL:
(www.ebi.ac.uk)
The EMBL Nucleotide Sequence Database
The EMBL
Nucleotide Sequence Database is a comprehensive database of DNA and RNA
sequences collected from the scientific literature and patent applications and
directly submitted from researchers and sequencing groups. Data collection is
done in collaboration with GenBank (USA) and the DNA Database of Japan (DDBJ).
The database currently doubles in size approximately every 12 months and
currently (December 1997) contains over 1,281 million bases from 1,917,868
sequence entries.
The complete
database is available every three months via subscription on CD-ROM. Computer
network services additionally provide access to the very latest data as well as
sequence similarity searches.
Documentation
·
Release Notes
·
Release 53,
December 1997.
·
Release 52,
October 1997.
·
Release 51,
June 1997.
·
Release 50,
March 1997.
·
Release 49,
December 1996.
·
Release 48,
September 1996.
·
Older releases
·
User Manual,
December 1997.
·
DDBJ/EMBL/GenBank
Feature Table Definition, v2.0, December 15, 1997.
Access
·
Subscription to
full releases on CD-ROM.
·
FTP archive
·
Full release (Release
53, December 1997).
·
Updates, daily, weekly
and cumulative.
·
Entry retrieval by database
accession number.
·
E-mail access to
data and sequence search services.
·
EMBnet nodes.
<!-- hhmts start -->Last modified: Thu January 08 1998 17:42
Growth of EMBL
database

EMBL and Genbank formats
EMBL format
ID LISOD standard; DNA; PRO; 756 BP.
XX
AC X64011;
S78972;
XX
NI g44010
XX
DT 28-APR-1992
(Rel. 31, Created)
DT 30-JUN-1993
(Rel. 36, Last updated, Version 6)
XX
DE L.ivanovii
sod gene for superoxide dismutase
XX.
KW sod gene;
superoxide dismutase.
XX
OS Listeria
ivanovii
OC Eubacteria;
Firmicutes; Low G+C gram-positive bacteria; Bacillaceae;
OC Listeria.
XX
RN [1]
RA Haas A.,
Goebel W.;
RT "Cloning
of a superoxide dismutase gene from Listeria ivanovii by
RT functional
complementation in Escherichia coli and
RT
characterization of the gene product.";
RL Mol. Gen.
Genet. 231:313-322(1992).
XX
RN [2]
RP 1-756
RA Kreft J.;
RT ;
RL Submitted
(21-APR-1992) on tape to the EMBL Data Library by:
RL J. Kreft,
Institut f. Mikrobiologie, Universitaet Wuerzburg,
RL Biozentrum Am
Hubland, 8700 Wuerzburg, FRG
XX
DR SWISS-PROT;
P28763; SODM_LISIV.
XX
FH Key Location/Qualifiers
FH
FT source 1..756
FT
/organism="Listeria ivanovii"
FT
/strain="ATCC 19119"
FT RBS 95..100
FT
/gene="sod"
FT CDS 109..717
FT
/gene="sod"
FT
/EC_number="1.15.1.1"
FT
/product="superoxide dismutase"
FT
/db_xref="PID:g44011"
FT
/db_xref="SWISS-PROT:P28763"
FT
/translation="MTYELPKLPYTYDALEPNFDKETMEIHYTKHHNIYVTKLNEAV
FT
SGHAELASKPGEELVANLDSVPEEIRGAVRNHGGGHANHTLFWSSLSPNGGGAPTGN
FT
LKAAIESEFGTFDEFKEKFNAAAAARFGSGWAWLVVNNGKLEIVSTANQDSPLSEGK
FT
TPVLGLDVWEHAYYLKFQNRRPEYIDTFWNVINWDERNKRFDAAK*"
FT
terminator 723..746
FT
/gene="sod"
XX
SQ Sequence 756
BP; 247 A; 136 C; 151 G; 222 T; 0 other;
CGTTATTTAA GGTGTTACAT AGTTCTATGG AAATAGGGTC TATACCTTTC
GCCTTACAAT 60
GTAATTTCTT TTCACATAAA TAATAAACAA TCCGAGGAGG AATTTTTAAT
GACTTACGAA 120
TTACCAAAAT TACCTTATAC TTATGATGCT TTGGAGCCGA ATTTTGATAA
AGAAACAATG 180
GAAATTCACT ATACAAAGCA CCACAATATT TATGTAACAA AACTAAATGA
AGCAGTCTCA 240
GGACACGCAG AACTTGCAAG TAAACCTGGG GAAGAATTAG TTGCTAATCT
AGATAGCGTT 300
CCTGAAGAAA TTCGTGGCGC AGTACGTAAC CACGGTGGTG GACATGCTAA
CCATACTTTA 360
TTCTGGTCTA GTCTTAGCCC AAATGGTGGT GGTGCTCCAA CTGGTAACTT
AAAAGCAGCA 420
ATCGAAAGCG AATTCGGCAC ATTTGATGAA TTCAAAGAAA AATTCAATGC
GGCAGCTGCG 480
GCTCGTTTTG GTTCAGGATG GGCATGGCTA GTAGTGAACA ATGGTAAACT
AGAAATTGTT 540
TCCACTGCTA ACCAAGATTC TCCACTTAGC GAAGGTAAAA CTCCAGTTCT
TGGCTTAGAT 600
GTTTGGGAAC ATGCTTATTA TCTTAAATTC CAAAACCGTC GTCCTGAATA
CATTGACACA 660
TTTTGGAATG TAATTAACTG GGATGAACGA AATAAACGCT TTGACGCAGC
AAAATAATTA 720
TCGAAAGGCT CACTTAGGTG GGTCTTTTTA TTTCTA 756
GenBank Format
LOCUS
LISOD 756 bp DNA BCT
30-JUN-1993
DEFINITION
L.ivanovii sod gene for superoxide dismutase.
ACCESSION X64011
S78972
NID g44010
KEYWORDS sod
gene; superoxide dismutase.
SOURCE Listeria ivanovii.
ORGANISM Listeria ivanovii
Eubacteria; Firmicutes; Low G+C gram-positive bacteria;
Bacillaceae; Listeria.
REFERENCE 1 (bases 1 to 756)
AUTHORS Haas,A. and Goebel,W.
TITLE Cloning of a superoxide dismutase gene
from Listeria ivanovii by
functional complementation in Escherichia coli and characterization
of the
gene product
JOURNAL Mol. Gen. Genet. 231 (2), 313-322 (1992)
MEDLINE 92140371
REFERENCE 2 (bases 1 to 756)
AUTHORS Kreft,J.
TITLE Direct Submission
JOURNAL Submitted (21-APR-1992) J. Kreft, Institut
f. Mikrobiologie,
Universitaet Wuerzburg, Biozentrum Am Hubland, 8700 Wuerzburg, FRG
FEATURES
Location/Qualifiers
source 1..756
/organism="Listeria ivanovii"
/strain="ATCC 19119"
/db_xref="taxon:1638"
RBS 95..100
/gene="sod"
gene 95..746
/gene="sod"
CDS 109..717
/gene="sod"
/EC_number="1.15.1.1"
/codon_start=1
/product="superoxide dismutase"
/db_xref="PID:g44011"
/db_xref="SWISS-PROT:P28763"
/transl_table=11
/translation="MTYELPKLPYTYDALEPNFDKETMEIHYTKHHNIYVTKLNEAVS
GHAELASKPGEELVANLDSVPEEIRGAVRNHGGGHANHTLFWSSLSPNGGGAPTGNLK
AAIESEFGTFDEFKEKFNAAAAARFGSGWAWLVVNNGKLEIVSTANQDSPLSEGKTPV
LGLDVWEHAYYLKFQNRRPEYIDTFWNVINWDERNKRFDAAK"
terminator 723..746
/gene="sod"
BASE COUNT
247 a 136 c 151 g
222 t
ORIGIN
1 cgttatttaa
ggtgttacat agttctatgg aaatagggtc tatacctttc gccttacaat
61
gtaatttctt ttcacataaa taataaacaa tccgaggagg aatttttaat gacttacgaa
121
ttaccaaaat taccttatac ttatgatgct ttggagccga attttgataa agaaacaatg
181
gaaattcact atacaaagca ccacaatatt tatgtaacaa aactaaatga agcagtctca
241
ggacacgcag aacttgcaag taaacctggg gaagaattag ttgctaatct agatagcgtt
301
cctgaagaaa ttcgtggcgc agtacgtaac cacggtggtg gacatgctaa ccatacttta
361
ttctggtcta gtcttagccc aaatggtggt ggtgctccaa ctggtaactt aaaagcagca
421
atcgaaagcg aattcggcac atttgatgaa ttcaaagaaa aattcaatgc ggcagctgcg
481
gctcgttttg gttcaggatg ggcatggcta gtagtgaaca atggtaaact agaaattgtt
541
tccactgcta accaagattc tccacttagc gaaggtaaaa ctccagttct tggcttagat
601
gtttgggaac atgcttatta tcttaaattc caaaaccgtc gtcctgaata cattgacaca
661
ttttggaatg taattaactg ggatgaacga aataaacgct ttgacgcagc aaaataatta
721
tcgaaaggct cacttaggtg ggtcttttta tttcta
//
FORMAT OF THE EMBL DATABASE
The nucleotide
sequence database is composed of sequence entries. Each entry corresponds to a
single contiguous sequence as contributed or reported in the literature. In
many cases, entries have been assembled from several papers reporting
overlapping sequence regions. Conversely, a single paper often provides data
for several entries, as when homologous sequences from different organisms are
compared.
·
3.1 Classes of Data
·
3.2 Database Divisions
·
3.3 Structure of an
Entry
·
3.4 Line Structure
·
3.4.1 The ID Line
·
3.4.2 The AC Line
·
3.4.3 The NI Line
·
3.4.4 The DT Line
·
3.4.5 The DE Line
·
3.4.6 The KW Line
·
3.4.7 The OS Line
·
3.4.8 The OC Line
·
3.4.9 The OG Line
·
3.4.10 The Reference (RN, RC,
RP, RX, RA, RT, RL) Lines
·
3.4.10.1 The RN Line
·
3.4.10.2 The RC Line
·
3.4.10.3 The RP Line
·
3.4.10.4 The RX Line
·
3.4.10.5 The RA Line
·
3.4.10.6 The RT Line
·
3.4.10.7 The RL Line
·
3.4.11 The DR Line
·
3.4.12 The FH Line
·
3.4.13 The FT Line
·
3.4.14 The SQ Line
·
3.4.15 The Sequence Data
Line
·
3.4.16 The CC Line
·
3.4.17 The XX Line
·
3.4.18 The // Line
<!-- hhmts end -->
3.2.4 Feature key examples
Key
Description
conflict
Separate determinations of the "same" sequence differ
rep_origin
Origin of replication
protein_bind
Protein binding site on DNA
CDS
Protein-coding sequence
misc_RNA
Generic label for an undefined RNA
insertion_seq
Insertion element
D-loop
Mitochondrial or other D-loop structure
3.3.4 Qualifier examples
Key Location/Qualifiers
CDS 86..742
/product="hypoxanthine
phosphoribosyltransferase"
/label=hprt
/note="hprt catalyzes vital steps
in the
reutilization pathway for purine
biosynthesis
and its deficiency leads to forms of
""gouty"" arthritis"
rep.origin 234..243
/direction=left
CDS 109..564
/usedin=X10009:catalase
3.5.3 Location examples
The following is a list of common location descriptors
with their meanings:
Location Description
467 Points to a single base in the
presented sequence
340..565 Points to a continuous range
of bases bounded by and including the
starting and ending bases
<345..500
Indicates that the exact
lower boundary point of a
feature is unknown. The location begins at some
base previous to the first base
specified (which need
not be contained in the presented
sequence) and con-
tinues to and includes the ending
base
<1..888
The feature starts
before the first sequenced base and
continues to and includes base 888
(102.110) Indicates that the exact location is
unknown but that
it is one of the bases between bases
102 and 110, in-
clusive
(23.45)..600 Specifies that the starting point is
one of the bases between bases 23 and 45, inclusive, and the end point is base
600
(122.133)..(204.221) The feature
starts at a base between 122 and 133, inclusive, and ends at a base between 204
and 221, inclusive
123^124 Points to a site between bases 123
and 124
145^177 Points to a site between two adjacent
bases anywhere
between bases 145 and 177
complement(34..(122.126)) Start at
one of the bases complementary to those between 122 and 126 on the presented
strand and finish at the base complementary to base 34 (the feature is on the
strand complementary to the presented strand)
join("acct",449..670) Concatenate the four bases 'acct' to the 5' end of the
sequence from bases 449 to 670,
inclusive
J00193:hladr Points to a feature whose location is
described in an-
other entry: the feature
labelled 'hladr' in the entry (in this database) with primary accession number
'J00193'
J00194:(100..202) Points to bases 100 to 202, inclusive, in the entry (in
this database) with primary accession
number
'J00194'
EMBL divisions
1 RELEASE 53
The EMBL Nucleotide Sequence Database was frozen to make Release 53 on the
16th December 1997. The release contains 1,917,868 sequence entries comprising
1,281,391,651 nucleotides. This represents an increase of about 8% over Release
52. A breakdown of Release 53 by division is shown below:
Division Entries Nucleotides
----------------- ------- -----------
Bacteriophage 1388 2188305
ESTs 1343796 496603984
Fungi 18137 44602064
GSSs 100154 49099107
HTG 1868 102763872
Human 74384 139022655
Invertebrates 28126 107524431
Organelles 24715 22870076
Other Mammals 14429 13785092
Other
Vertebrates 13145 14653255
Plants 22136 37736590
Patent 91221 29511807
Prokaryotes 42666 102750354
Rodents 37043 46489741
STSs 51172 17685717
Synthetic 2424 5377292
Unclassified 2380 2387088
Viruses 48684 46340221
----------------- --------
------------
Total 1917868 1281391651
----------------- -------- ------------
hum Human Sequences | |
rod RodentSequences |>Mammals
|
mam Other Mammal Sequences |
|> Vertebrates
vrt Other Vertebrate Sequences |
STS (Sequence
Tagged Sites)
Sequence Tagged
Sites (STS) are short DNA segments with a single location in the
genome. This
feature of STS makes them useful tags for mapping.
EST (Expressed
Sequence Tag)
Description
Expressed Sequence Tags (ESTs) are sequences of cDNA which have been
reverse-transcribed
from mRNA and their function is not necessarily known. They have
applications in
the discovery of new genes, mapping of various genomes, and identification of
coding regions in
genomic sequences.
Genome Survey
Sequence (GSS)
Genome Survey
Sequence, includes single-pass genomic data, exon-trapped sequences, and Alu
PCR sequences.
dbGSS release
030598 - Summary by Organism - March 5, 1998
Number
of public entries: 88,416
Homo
sapiens (human) 64,533
Arabidopsis
thaliana 22,511
Trypanosoma
brucei brucei 455
Trypanosoma
brucei rhodesiense 324
Cryptosporidium
parvum 200
Mus
musculus 163
Rhodobacter
sphaeroides 151
Enterococcus
faecalis 41
Helicobacter
pylori 21
Brugia
malayi 11
Leishmania
major 6
High-Throughput
Genomic Sequences
The High
Throughput Genomic (HTG) Sequences division was created to accommodate a
growing need to make 'unfinished' genomic sequence data rapidly available to
the scientific community. It was done in a coordinated effort between the three
International Nucleotide Sequence databases: DDBJ, EMBL, and GenBank. The HTG
division contains 'unfinished' DNA sequences generated by the high-throughput
sequencing centers. Sequence data in this division are available for BLAST
homology searches against either the "htgs" database or the
"month" database, which includes all new submissions for the prior
month. The HTG division of GenBank was described in a [Genome Research (1997) 7(10)]
article by Ouellette and Boguski.
Location of HTG
records:
Unfinished HTG
sequences containing contigs greater than 2 kb are
assigned an
accession number and deposited in the HTG division. A typical
HTG record might
consist of all the first pass sequence data generated from
a single cosmid,
BAC, YAC, or P1 clone which together comprise more
than 2 kb and
contain one or more gaps. A single accession number is
assigned to this
collection of sequences and each record includes a clear
indication of the
status (phase 1 or 2) plus a prominent warning that the
sequence data is
"unfinished" and may contain errors. The accession
number does not
change as sequence records are updated; only the most
recent version of
a HTG record remains in GenBank. 'Finished' HTG
sequences (phase
3) retain the same accession number, but are moved into
the relevant
primary GenBank division. An example of a submission (one
accession number)
that has progressed through phase 1, phase 2, and
phase 3 is
available
TIGR Microbial
Database
A listing of microbial genomes that have been
published or are in the process of being sequenced.
Published microbial genomes
|
|
Link
|
Genome
|
Strain
|
Domain
|
Size (Mb)
|
Institution
|
Funding
|
Publication
|
|
1
|

|
Haemophilus
influenzae Rd
|
KW20
|
B
|
1.83
|
TIGR
|
TIGR
|
Fleischmann
et. al., Science 269:496-512 (1995)
|
|
2
|

|
Mycoplasma
genitalium
|
G-37
|
B
|
0.58
|
TIGR
|
DOE
|
Fraser
et. al., Science 270:397-403 (1995)
|
|
3
|

|
Methanococcus jannaschii
|
DSM 2661
|
A
|
1.66
|
TIGR
|
DOE
|
Bult
et. al., Science 273:1058-1073 (1996)
|
|
4
|

|
Synechocystis sp.
|
PCC 6803
|
B
|
3.57
|
Kazusa DNA Research
Inst.
|
|
Kaneko
et. al., DNA Res. 3: 109-136 (1996)
|
|
5
|

|
Mycoplasma pneumoniae
|
M129
|
B
|
0.81
|
Univ.
of Heidelberg
|
|
Himmelreich
et. al., Nuc. Acid Res. 24:4420-4449 (1996)
|
|
6
|


|
Saccharomyces cerevisiae
|
S288C
|
E
|
13
|
International Consortium
|
EC, NHGRI, Welcome Trust, McGill U., RIKEN
|
Goffeau
et. al., Nature 387 (Suppl.) 5-105 (1997)
|
|
7
|

|
Helicobacter pylori
|
26695
|
B
|
1.66
|
TIGR
|
TIGR
|
Tomb
et. al., Nature388:539-547 (1997)
|
|
8
|

|
Escherichia coli
|
K-12
|
B
|
4.60
|
University
of Wisconsin
|
NHGRI
|
Blattner
et. al., Science 277:1453-1474 (1997)
|
|
9
|

|
Methanobacterium thermoautotrophicum
|
delta H
|
A
|
1.75
|
Genome
Therapeutics & Ohio State Univ.
|
DOE
|
Smith
et.al., J. Bacteriology, 179:7135-7155 (1997)
|
|
10
|

|
Bacillus subtilis
|
168
|
B
|
4.20
|
International Consortium
|
EC
|
Kunst
et.al., Nature390: 249-256 (1997)
|
|
11
|

|
Archaeoglobus fulgidus
|
VC-16, DSM4304
|
A
|
2.18
|
TIGR
|
DOE
|
Klenk
et al.,Nature 390:364-370 (1997)
|
|
12
|

|
Borrelia
burgdorferi
|
B31
|
B
|
1.44
|
TIGR
|
Mathers Foundation
|
Fraser
et al., Nature, 390: 580-586 (1997)
|

Microbial genomes in progress
KEY
|
Domain
|
A: Archaea
|
B: Eubacteria
|
E: Eucaryote
|

Map of Mycoplasma genitalium genome.
The flow of genetic information
DNA -> RNA -> protein -> conformation
Translation products of DNA - Amino acids in three letter code
ValArgIleArgIleSerAsp
TyrGlyPheGlyPheArgMet
ThrAspSerAspPheGlyCys
5' GUACGGAUUCGGAUUUCGGAUGC
3'
3' CAUGCCUAAGCCUAAAGCCUACG
5'
TyrProAsnProAsnArgIle
ValSerGluSerLysProHis
ArgIleArgIleGluSerAla
Amino acids in one letter code
V R
I R I S D
Y G
F G F R M
T D
S D F G C
5' GUACGGAUUCGGAUUUCGGAUGC 3'
3' CAUGCCUAAGCCUAAAGCCUACG 5'
Y P
N P N R I
V S
E S K P H
R I
R I E S A
Three- and one-letter codes of the amino acids.
Alanine Ala A
Arginine Arg R
Asparagine Asn N
Aspartate Asp D
Cysteine Cys C
Glutamate Glu E
Glutamine Gln Q
Glycine Gly G
Histidine His H
Isoleucine Ile I
Leucine Leu L
Lysine Lys K
Metionine Met M
Fenylalanine Phe F
Proline Pro P
Serine Ser S
Treonine Thr T
Tryptofan Trp W
Tyrosine Tyr Y
Valine Val V
4. THE GENETIC
CODE
UUU Phe UCU Ser
UAU Tyr UGU
Cys
UUC Phe UCC Ser
UAC Tyr UGC
Cys
UUA Leu UCA Ser
UAA Stop UGA
Stop
UUG Leu UCG Ser
UAG Stop UGG
Trp

CUU Leu CCU Pro
CAU His CGU
Arg
CUC Leu CCC Pro
CAC His CGC
Arg
CUA Leu CCA Pro
CAA Gln CGA
Arg
CUG Leu CCG Pro
CAG Gln CGG
Arg

AUU Ile ACU Thr
AAU Asn AGU
Ser
AUC Ile ACC Thr
AAC Asn AGC
Ser
AUA Ile ACA Thr
AAA Lys AGA
Arg
AUG Met ACG Thr
AAG Lys AGG
Arg

GUU Val GCU Ala
GAU Asp GGU
Gly
GUC Val GCC Ala
GAC Asp GGC
Gly
GUA Val GCA Ala
GAA Glu GGA
Gly
GUG Val GCG Ala
GAG Glu GGG
Gly

Table I. The genetic code
Sequence
symbols: Nucleotides
Symbol Meaning Complement
A A T
C C G
G G C
T/U T A
M A or C K
R A or G Y
W A or T W
S C or G S
Y C or T R
K G or T M
V A or C or G B
H A or C or T D
D A or G or T H
B C or G or T V
X/N G or A or T or C X
. not G or A or T or C .
Deviations from the standard genetic code
# Cilian protozoa
UAA =
Gln:Q
UAG =
Gln:Q
# Yeast mitochondria
UGA =
Trp:W
CUU =
Thr:T
CUC =
Thr:T
CUA =
Thr:T
CUG =
Thr:T
AUA =
Met:M
# Mammalian mitochondria
UGA =
Trp:W
AUU =
Ile:I
AUC =
Ile:I
AUA =
Met:M
AGA = * :*
AGG = * :*
# Drosophila mitochondria
UGA =
Trp:W
AUU =
Ile:I
AUA =
Met:M
AGA =
Ser:S
AGG =
Ser:S
# mycoplasma
UGA = Trp
Codon usage for enteric bacterial (highly expressed) genes 7/19/83
AmAcid Codon Number
/1000 Fraction ..
Gly GGG 13.00 1.89 0.02
Gly GGA 3.00 0.44 0.00
Gly GGU 365.00 52.99 0.59
Gly GGC 238.00 34.55 0.38
Glu GAG 108.00 15.68 0.22
Glu GAA 394.00 57.20 0.78
Asp GAU 149.00 21.63 0.33
Asp GAC 298.00 43.26 0.67
Val GUG 93.00 13.50 0.16
Val GUA 146.00 21.20 0.26
Val GUU 289.00 41.96 0.51
Val GUC 38.00 5.52 0.07
Ala GCG 161.00 23.37 0.26
Ala GCA 173.00 25.12 0.28
Ala GCU 212.00 30.78 0.35
Ala GCC 62.00 9.00 0.10
Arg AGG 1.00 0.15 0.00
Arg AGA 0.00 0.00 0.00
Ser AGU 9.00 1.31 0.03
Ser AGC 71.00 10.31 0.20
Lys AAG 111.00 16.11 0.26
Lys AAA 320.00 46.46 0.74
Asn AAU 19.00 2.76 0.06
Asn AAC 274.00 39.78 0.94
Met AUG 170.00 24.68 1.00
Ile AUA 1.00 0.15 0.00
Ile AUU 70.00 10.16 0.17
Ile AUC 345.00 50.09 0.83
Thr ACG 25.00 3.63 0.07
Thr ACA 14.00 2.03 0.04
Thr ACU 130.00 18.87 0.35
Thr ACC 206.00 29.91 0.55
Trp UGG 55.00 7.98 1.00
End UGA 0.00 0.00 0.00
Cys UGU 22.00 3.19 0.49
Cys UGC 23.00 3.34 0.51
End UAG 0.00 0.00 0.00
End UAA 0.00 0.00 0.00
Tyr UAU 51.00 7.40 0.25
Tyr UAC 157.00 22.79 0.75
Leu UUG 18.00 2.61 0.03
Leu UUA 12.00 1.74 0.02
Phe UUU 51.00 7.40 0.24
Phe UUC 166.00 24.10 0.76
Ser UCG 14.00 2.03 0.04
Ser UCA 7.00 1.02 0.02
Ser UCU 120.00 17.42 0.34
Ser UCC 131.00 19.02 0.37
Arg CGG 1.00 0.15 0.00
Arg CGA 2.00 0.29 0.01
Arg CGU 290.00 42.10 0.74
Arg CGC 96.00 13.94 0.25
Gln CAG 233.00 33.83 0.86
Gln CAA 37.00 5.37 0.14
His CAU 18.00 2.61 0.17
His CAC 85.00 12.34 0.83
Leu CUG 480.00 69.69 0.83
Leu CUA 2.00 0.29 0.00
Leu CUU 25.00 3.63 0.04
Leu CUC 38.00 5.52 0.07
Pro CCG 190.00 27.58 0.77
Pro CCA 36.00 5.23 0.15
Pro CCU 19.00 2.76 0.08
Pro CCC 1.00 0.15 0.00
Swissprot
The SWISS-PROT
Protein Sequence Data Bank is a database of protein sequences produced
collaboratively by Amos Bairoch (University of Geneva) and the EBI. Itcontains
high-quality annotation, is non-redundant, and cross-referenced to many other
databases.
Release 35.0 of
SWISS-PROT contains 69'113 sequence entries, comprising 25'083'768 amino acids
abstracted from 59'101 references.
SWISS-PROT is
accompanied by TREMBL, a computer-annotated supplement to SWISS-PROT. TREMBL
contains the translations of all coding sequences (CDS) present in the EMBL
Nucleotide Sequence Database not yet integrated into SWISS-PROT. TREMBL can be
considered as a preliminary section of SWISS-PROT as all TREMBL entries have
been assigned SWISS-PROT accession numbers. TREMBL is split into two main sections;
SP-TREMBL and REM-TREMBL. SP-TREMBL (SWISS-PROT TREMBL) contains the entries
which should eventually be incorporated into SWISS-PROT.
Release 5 of
TREMBL is created from release 53 of the EMBL nucleotide sequence database and
contains 166'361 sequence entries, comprising 45'671'684 amino acids.
General Documents
·
User Manual
·
Release Notes
·
Current SWISS-PROT release
·
Old SWISS-PROT release notes
·
Current TREMBL release
notes
·
Old TREMBL release
notes
·
SWISS-PROT Documentation
·
Publications of the EBI
SWISS-PROT Team.
·
How to contact
SWISS-PROT
Swissprot entry
ID
PRIO_HUMAN STANDARD; PRT;
253 AA.
AC P04156;
DT 01-NOV-1986
(REL. 03, CREATED)
DT 01-NOV-1986
(REL. 03, LAST SEQUENCE UPDATE)
DT 01-NOV-1997
(REL. 35, LAST ANNOTATION UPDATE)
DE MAJOR PRION
PROTEIN PRECURSOR (PRP) (PRP27-30) (PRP33-35C) (ASCR).
GN PRNP.
OS HOMO SAPIENS
(HUMAN).
OC EUKARYOTA;
METAZOA; CHORDATA; VERTEBRATA; TETRAPODA; MAMMALIA;
OC EUTHERIA;
PRIMATES.
RN [1]
RP SEQUENCE FROM
N.A.
RX MEDLINE;
86300093.
RA KRETZSCHMAR
H.A., STOWRING L.E., WESTAWAY D., STUBBLEBINE W.H.,
RA PRUSINER
S.B., DEARMOND S.J.;
RL DNA 5:315-324(1986).
RN [2]
RP SEQUENCE OF
8-253 FROM N.A.
RX MEDLINE;
86261778.
RA LIAO Y.-C.J.,
LEBO R.V., CLAWSON G.A., SMUCKLER E.A.;
RL SCIENCE
233:364-367(1986).
RN [3]
RP VARIANT
AMYLOID GSS, SEQUENCE OF 58-85 AND 111-150.
RX MEDLINE;
91160504.
RA TAGLIAVINI
F., PRELLI F., GHISO J., BUGIANI O., SERBAN D.,
RA PRUSINER
S.B., FARLOW M.R., GHETTI B., FRANGIONE B.;
RL EMBO J.
10:513-519(1991).
RN [4]
RP REVIEW ON
VARIANTS.
RX MEDLINE;
93372867.
RA PALMER M.S.,
COLLINGE J.;
RL HUM. MUTAT.
2:168-173(1993).
RN [5]
RP REVIEW ON
VARIANTS.
RX MEDLINE;
94029646.
RA PRUSINER
S.B.;
RL ARCH. NEUROL.
50:1129-1153(1993).
RN [6]
RP VARIANT GSS
LEU-102.
RX MEDLINE;
89159432.
RA HSIAO K.,
BAKER H.F., CROW T.J., POULTER M., OWEN F.,
RA TERWILLIGER
J.D., WESTAWAY D., OTT J., PURSINER S.B.;
RL NATURE
338:342-345(1989).
RN [7]
RP VARIANTS
LEU-102; VAL-117 AND VAL-129.
RX MEDLINE;
89392018.
RA DOH-URA K.,
TATEISHI J., SASAKI H., KITAMOTO T., SAKAKI Y.;
RL BIOCHEM.
BIOPHYS. RES. COMMUN. 163:974-979(1989).
RN [8]
RP VARIANT FFI
ASN-178.
RX MEDLINE;
92195483.
RA MEDORI R.,
MONTAGNA P., TRITSCHLER H.J., LEBLANC A., CORTELLI P.,
RA TINUPER P.,
LUGARESI E., GAMBETTI P.;
RL NEUROLOGY
42:669-670(1992).
RN [9]
RP VARIANT CJD
ASN-178.
RX MEDLINE;
91124933.
RA GOLDFARB
L.G., HALTIA M., BROWN P., NIETO A., KOVANEN J.,
RA MCCOMBIE
W.R., TRAPP S., GAJDUSEK D.C.;
RL LANCET
337:425-425(1991).
RN [10]
RP VARIANT CJD
LYS-200.
RX MEDLINE;
90355709.
RA GOLDFARB L.,
MITROVA E., BROWN P., TOH B.K., GAJDUSEK D.C.;
RL LANCET
336:514-515(1990).
RN [11]
RP VARIANT GSS
ARG-217.
RX MEDLINE;
93250977.
RA HSIAO K.,
DLOUHY S.R., FARLOW M.R., CASS C., DA COSTA M.,
RA CONNEALLY
P.M., HODES M.E., GHETTI B., PRUSINER S.B.;
RL NAT. GENET.
1:68-71(1992).
RN [12]
RP VARIANTS CJD
ILE-180 AND ARG-223.
RX MEDLINE;
93213314.
RA KITAMOTO T.,
OHTA M., DOH-URA K., HITOSHI S., TERAO Y., TATEISHI J.;
RL BIOCHEM.
BIOPHYS. RES. COMMUN. 191:709-714(1993).
RN [13]
RP VARIANT CJD
ILE-210.
RX MEDLINE;
94071412.
RA POCCHIARI M.,
SALVATORE M., CUTRUZZOLA F., GENUARDI M.,
RA ALLCATELLI
C.T., MASULLO C., MACCHI G., ALEMA G., GALGANI S., XI Y.G.,
RA PETRAROLI R.,
SILVESTRINI M.C., BRUNORI M.;
RL ANN. NEUROL.
34:802-807(1993).
RN [14]
RP VARIANT GSS
LEU-105.
RX MEDLINE;
94077414.
RA YAMADA M.,
ITOH Y., FUJIGASAKI H., NARUSE S., KANEKO K., KITAMOTO T.,
RA TATEISHI J.,
OTOMO E., HAYAKAWA M., TANAKA J., MATSUSHITA M.,
RA MIYATAKE T.;
RL NEUROLOGY
43:2723-2724(1993).
RN [15]
RP VARIANT GSS
LEU-105.
RX MEDLINE;
95213742.
RA ITOH Y.,
YAMADA M., HAYAKAWA M., SHOZAWA T., TANAKA J., MATSUSHITA M.,
RA KITAMOTO T.,
TATEISHI J., OTOMO E.;
RL J. NEUROL.
SCI. 127:77-86(1994).
RN [16]
RP VARIANT CJD
LYS-200.
RX MEDLINE;
94142912.
RA INOUE I.,
KITAMOTO T., DOH-URA K., SHII H., GOTO I., TATEISHI J.;
RL NEUROLOGY
44:299-301(1994).
RN [17]
RP VARIANT CJD
LYS-200.
RX MEDLINE;
94316708.
RA GABIZON R.,
ROSENMAN H., MEINER Z., KAHANA I., KAHANA E., SHUGART Y.,
RA OTT J.,
PRUSINER S.B.;
RL PHILOS.
TRANS. R. SOC. LOND., B, BIOL. SCI. 343:385-390(1994).
RN [18]
RP VARIANT GSS
LEU-102.
RX MEDLINE;
95303274.
RA YOUNG K.,
JONES C.K., PICCARDO P., LAZZARINI A., GOLBE L.I.,
RA ZIMMERMAN
T.R., DICKSON D.W., MCLACHLAN D.C., ST GEORGE-HYSLOP P.,
RA LENNOX A.;
RL NEUROLOGY
45:1127-1134(1995).
CC -!- FUNCTION:
THE FUNCTION OF PRP IS NOT KNOWN. PRP IS ENCODED IN THE
CC HOST
GENOME AND IS EXPRESSED BOTH IN NORMAL AND INFECTED CELLS.
CC -!- SUBUNIT:
PRP HAS A TENDENCY TO AGGREGATE YIELDING POLYMERS CALLED
CC
"RODS".
CC -!-
SUBCELLULAR LOCATION: ATTACHED TO THE MEMBRANE BY A GPI-ANCHOR.
CC -!- DISEASE:
PRP IS FOUND IN HIGH QUANTITY IN THE BRAIN OF HUMANS AND
CC ANIMALS
INFECTED WITH NEURODEGENERATIVE DISEASES KNOWN AS
CC
TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES OR PRION DISEASES, LIKE:
CC
CREUTZFELDT-JACOB DISEASE (CJD), GERSTMANN-STRAUSSLER SYNDROME
CC (GSS),
FATAL FAMILIAL INSOMNIA (FFI) AND KURU IN HUMANS; SCRAPIE
CC IN SHEEP
AND GOAT; BOVINE SPONGIFORM ENCEPHALOPATHY (BSE) IN
CC CATTLE;
TRANSMISSIBLE MINK ENCEPHALOPATHY (TME); CHRONIC WASTING
CC DISEASE
(CWD) OF MULE DEER AND ELK; FELINE SPONGIFORM
CC
ENCEPHALOPATHY (FSE) IN CATS AND EXOTIC UNGULATE ENCEPHALOPATHY
CC (EUE) IN
NYALA AND GREATER KUDU. THE PRION DISEASES ILLUSTRATE
CC THREE
MANIFESTATIONS OF CNS DEGENERATION: (1) INFECTIOUS (2)
CC SPORADIC
AND (3) DOMINANTLY INHERITED FORMS. TME, CWD, BSE, FSE,
CC EUE ARE
ALL THOUGHT TO OCCUR AFTER CONSUMPTION OF PRION-INFECTED
CC
FOODSTUFFS.
CC -!- DISEASE:
CJD OCCURS PRIMARILY AS A SPORADIC DISORDER (1 PER
CC MILLION),
WHILE 10-15% ARE FAMILIAL. ACCIDENTAL TRANSMISSION OF
CC CJD TO
HUMANS APPEARS TO BE IATROGENIC (CONTAMINATED HUMAN GROWTH
CC HORMONE
(HGH), CORNEAL TRANSPLANTATION, ELECTROENCEPHALOGRAPHIC
CC ELECTRODE
IMPLANTATION. . .). EPIDEMIOLOGIC STUDIES HAVE FAILED TO
CC IMPLICATE
THE INGESTION OF INFECTED ANNIMAL MEAT IN THE
CC
PATHOGENESIS OF CJD IN HUMAN. THE TRIAD OF MICROSCOPIC FEATURES
CC THAT
CHARACTERIZE THE PRION DISEASES CONSISTS OF (1) SPONGIFORM
CC
DEGENERATION OF NEURONS, (2) SEVERE ASTROCYTIC GLIOSIS THAT OFTEN
CC APPEARS
TO BE OUT OF PROPORTION TO THE DEGREE OF NERF CELL LOSS,
CC AND (3)
AMYLOID PLAQUE FORMATION. CJD IS CHARACTERIZED BY
CC
PROGRESSIVE DEMENTIA AND MYOCLONIC SEIZURES, AFFECTING ADULTS IN
CC MID-LIFE.
SOME PATIENTS PRESENT SLEEP DISORDERS, ABNORMALITIES OF
CC HIGH
CORTICAL FUNCTION, CEREBELLAR AND CORTICOSPINAL DISTURBANCES.
CC THE
DISEASE ENDS IN DEATH AFTER A 3-12 MONTHS ILLNESS.
CC -!- DISEASE:
GSS IS A HETEROGENEOUS DISORDER AND WAS DEFINED AS A
CC
"SPINOCEREBELLAR ATAXIA WITH DEMENTIA AND PLAQUELIKE
DEPOSITS".
CC GSS
INCIDENCE IS LESS THAN 2 PER 100 MILLION.
CC -!- DISEASE:
KURU IS TRANSMITTED DURING RITUALISTIC CANNIBALISM, AMONG
CC NATIVES
OF THE NEW GUINEA HIGHLANDS. PATIENTS EXHIBIT VARIOUS
CC MOVEMENT
DISORDERS LIKE CEREBELLAR ABNORMALITIES, RIGIDITY OF THE
CC LIMBS,
AND CLONUS. EMOTIONNAL LABILITY IS PRESENT, AND DEMENTIA IS
CC CONSPICUOUSLY
ABSENT. DEATH USUALLY OCCURS FROM 3 TO 12 MONTH
CC AFTER
ONSET.
CC -!-
SIMILARITY: TO OTHER PRP.
CC -!- DATABASE:
NAME=HotMolecBase; NOTE=PrP entry;
CC
WWW="http://bioinformatics.weizmann.ac.il/hotmolecbase/entries/prp.htm".
DR EMBL; M13667;
G190470; -.
DR EMBL; M13899;
G190468; -.
DR EMBL; D00015;
G220016; -.
DR PIR; A05017;
A05017.
DR PIR; A24173;
A24173.
DR PIR; S14078;
S14078.
DR MIM; 176640;
-.
DR MIM; 123400;
-.
DR MIM; 137440;
-.
DR MIM; 245300;
-.
DR MIM; 600072;
-.
DR PROSITE;
PS00291; PRION_1; 1.
DR PROSITE;
PS00706; PRION_2; 1.
KW PRION; BRAIN;
GLYCOPROTEIN; GPI-ANCHOR; REPEAT; SIGNAL;
KW POLYMORPHISM;
DISEASE MUTATION.
FT SIGNAL 1
22
FT CHAIN 23
230 MAJOR PRION PROTEIN.
FT PROPEP 231
253 REMOVED IN MATURE FORM
(BY SIMILARITY).
FT LIPID 230
230 GPI-ANCHOR (BY
SIMILARITY).
FT CARBOHYD 181
181 PROBABLE.
FT CARBOHYD 197
197 PROBABLE.
FT DISULFID 179 214 BY SIMILARITY.
FT DOMAIN 51
91 5 X 8 AA TANDEM REPEATS
OF P-H-G-G-G-W-G-
FT Q.
FT REPEAT 51
59 1.
FT REPEAT 60
67 2.
FT REPEAT 68
75 3.
FT REPEAT 76
83 4.
FT REPEAT 84
91 5.
FT VARIANT 102
102 P -> L (IN GSS).
FT VARIANT 105
105 P -> L (IN GSS).
FT VARIANT 117
117 A -> V (LINKED TO
DEVELOPMENT OF
FT DEMENTING GSS).
FT VARIANT 129
129 M -> V (DETERMINES
THE DISEASE PHENOTYPE
FT IN PATIENTS WHO HAVE A PRP MUTATION
AT
FT CODON 178: PATIENTS WITH MET
DEVELOP FFI,
FT THOSE WITH VAL DEVELOP CJD).
FT VARIANT 178
178 D -> N (IN FFI AND
CJD).
FT VARIANT 180
180 V -> I (IN CJD).
FT VARIANT 198
198 F -> S (IN A
ATYPICAL FORM OF GSS WITH
FT NEUROFIBRILLARY TANGLES).
FT VARIANT 200
200 E -> K (IN CJD).
FT VARIANT 210
210 V -> I (IN CJD).
FT VARIANT 217
217 Q -> R (IN GSS WITH
NEUROFIBRILLARY
FT TANGLES).
FT VARIANT 232
232 M -> R (IN CJD).
FT CONFLICT 118
118 MISSING (IN REF. 2).
SQ SEQUENCE 253 AA;
27661 MW; FD5373AD CRC32;
MANLGCWMLV
LFVATWSDLG LCKKRPKPGG WNTGGSRYPG QGSPGGNRYP PQGGGGWGQP
HGGGWGQPHG
GGWGQPHGGG WGQPHGGGWG QGGGTHSQWN KPSKPKTNMK HMAGAAAAGA
VVGGLGGYML
GSAMSRPIIH FGSDYEDRYY RENMHRYPNQ VYYRPMDEYS NQNNFVHDCV
NITIKQHTVT
TTTKGENFTE TDVKMMERVV EQMCITQYER ESQAYYQRGS SMVLFSSPPV
ILLISFLIFL
IVG
//
PROSITE entries.
Example I
ID ATP_GTP_A;
PATTERN.
AC PS00017;
DT APR-1990
(CREATED); APR-1990 (DATA UPDATE); NOV-1990 (INFO UPDATE).
DE
ATP/GTP-binding site motif A (P-loop).
PA
[AG]-x(4)-G-K-[ST].
CC
/TAXO-RANGE=ABEPV;
3D 1EFM; 1ETU;
1Q21; 2Q21; 4Q21; 5Q21; 6Q21;
DO PDOC00017;
Example II
ID ZINC_FINGER_C2H2; PATTERN.
AC PS00028;
DT APR-1990 (CREATED); JUN-1994 (DATA UPDATE);
NOV-1997 (INFO UPDATE).
DE Zinc finger, C2H2 type, domain.
PA C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H.
NR /RELEASE=35,69113;
NR /TOTAL=1932(412); /POSITIVE=1891(372);
/UNKNOWN=6(6); /FALSE_POS=35(34);
NR /FALSE_NEG=3; /PARTIAL=1;
CC /TAXO-RANGE=??E?V; /MAX-REPEAT=37;
CC /SITE=1,zinc; /SITE=3,zinc; /SITE=7,zinc;
/SITE=9,zinc;
DR P21192, ACE2_YEAST, T; P07248, ADR1_YEAST,
T; P39413, AEF1_DROME, T;
DR Q00900, AGIE_RAT , T; P41696, AZF1_YEAST, T; Q01954, BASO_HUMAN, T;
DR P41182, BCL6_HUMAN, T; P41183, BCL6_MOUSE,
T; P55201, BR14_HUMAN, T;
DR Q01295, BRC1_DROME, T; Q01296, BRC2_DROME,
T; Q01293, BRC3_DROME, T;
DR P10069, BRLA_EMENI, T; Q01713,
BTEB_RAT , T; Q01522, CF23_DROME, T;
DR P20385, CF2_DROME , T; P19538, CID_DROME ,
T; Q05620, CREA_ASPNG, T;
DR Q01981, CREA_EMENI, T; Q08705, CTCF_CHICK,
T; P49711, CTCF_HUMAN, T;
DR P36197, DEFI_CHICK, T; P23792, DISC_DROME,
T; P26632, EGR1_BRARE, T;
DR P18146, EGR1_HUMAN, T; P08046, EGR1_MOUSE,
T; P08154, EGR1_RAT , T;
DR Q05159, EGR2_BRARE, T; P26633, EGR2_CRILO,
T; P26634, EGR2_DUSTH, T;
DR P11161, EGR2_HUMAN, T; P08152, EGR2_MOUSE,
T; P26635, EGR2_POERE, T;
DR P51774, EGR2_RAT , T; Q08427, EGR2_XENLA, T; Q06889, EGR3_HUMAN, T;
DR P43300, EGR3_MOUSE, T; P43301,
EGR3_RAT , T; Q05215, EGR4_HUMAN, T;
.
. ( I edited out a lot of very interesting
information here ...)
.
DR P49782, S3AE_BACSU, F; P24804, TA29_TOBAC,
F; P36810, VE6_HPV32 , F;
DR P15024, VL1_REOVD , F; P03527, VSI3_REOVD,
F; P30211, VSI3_REOVJ, F;
DR P07939, VSI3_REOVL, F; Q93098, WN8B_HUMAN,
F; P51028, WNT8_BRARE, F;
DR P28026, WNT8_XENLA, F; P20201, Y15K_SSV1 ,
F; P20198, Y5K6_SSV1 , F;
DR P43558, YFE4_YEAST, F; P37127, YFFG_ECOLI,
F; P38890, YH07_YEAST, F;
DR Q09441, YP83_CAEEL, F;
3D 1ARD; 1ARE; 1ARF; 1PAA; 1ZAA; 1AAY; 2GLI;
1SP1; 1SP2; 1NCS; 1ZFD; 1TF3;
3D 2DRP; 1ZNF; 3ZNF; 4ZNF; 1BBO; 5ZNF; 7ZNF;
DO PDOC00028;
PROSITE documentation
{PDOC00004}
{PS00004;
CAMP_PHOSPHO_SITE}
{BEGIN}
****************************************************************
* cAMP-
and cGMP-dependent protein kinase phosphorylation site *
****************************************************************
There has
been a number of studies relative to the specificity of cAMP- and
cGMP-dependent
protein kinases [1,2,3]. Both types of
kinases appear to share
a
preference for the
phosphorylation of serine or
threonine residues found
close to
at least two consecutive
N-terminal basic residues. It is
important
to note
that there are quite a number of exceptions to this rule.
-Consensus
pattern: [RK](2)-x-[ST]
[S or T is the
phosphorylation site]
-Last
update: June 1988 / First entry.
[ 1]
Fremisco J.R., Glass D.B., Krebs E.G.
J. Biol. Chem. 255:4240-4245(1980).
[ 2]
Glass D.B., Smith S.B.
J. Biol. Chem. 258:14797-14803(1983).
[ 3]
Glass D.B., El-Maghrabi M.R., Pilkis S.J.
J. Biol. Chem. 261:2987-2993(1986).
{END}
Enzyme
www.bis.med.jhmi.edu/Dan/proteins/ec-enzyme.html
The current release has 3650 entries and was indexed 01-Mar-1998.
Description
The ENZYME data
bank contains the following data for each type of
characterized
enzyme for which an EC number has been provided: EC
number,
Recommended name, Alternative names, Catalytic activity,
Cofactors,
Pointers to the SWISS-PROT entrie(s) that correspond to the
enzyme, Pointers
to disease(s) associated with a deficiency of the enzyme.
Literature
Bairoch A. (1993)
The ENZYME data bank. Nucleic Acids Res, Jul
1;21(13):3155-6.
Taxonomy database
www3.ncbi.nlm.nih.gov/Taxonomy/tax.html
This is the top level of the taxonomy database maintained by
NCBI/GenBank. You can explore any of the taxa listed below by clicking it.
Archaea
Eubacteria
Eukaryotae
Viroids
Viruses
Other
Unclassified
This is a searchable index. You can enter the name of superspecific taxa
(e.g., Porifera) or
the name of a particular organism (e.g., Thalarctos maritimus for the polar
bear or polar
bear itself).
Query:
Use query string as : Complete
match Regular expression Set of tokens
The "Set of tokens" option returns longer names that include the
search terms, e.g., hybrid taxa.
See what happens if you query "Bos taurus" using the
"Complete match" option versus the "Set of
tokens" option.
These are direct links to some of the organisms most commonly used in
molecular research projects:
Arabidopsis thaliana
Caenorhabditis elegans
Danio rerio (zebrafish)
Drosophila
Escherichia coli
Hepatitis C virus
Homo sapiens
Mus musculus
Mycoplasma
Oryza sativa
Plasmodium falciparum
Pneumocystis carinii
Rattus
Saccharomyces cerevisiae
Schizosaccharomyces pombe
Xenopus laevis
Molecular biology databases – text searches and retrieval of data.
With Internet tools such as
- Entrez
or
- Sequence Retrieval System
(SRS)
you can search the annotation section of molecular biology databases using
one or more words and by selecting specific fields in the database. For
instance to find all insulin proteins in a protein database you can search
Entrez – protein database , use ”insulin” as search word and selecting ”All
fields” or ”Protein Name”.
Tutorials for Entrez and SRS
http://www3.ncbi.nlm.nih.gov/Entrez/entrezhelp.html
http://srs.ebi.ac.uk:5000/srs5/man/srsman.html
Fields that may be specified in the nucleotide, protein and Pubmed databases of
Entrez:
SRS servers
|
WEHI, Melbourne, Australia
|
|
Belgian EMBnet Node (BEN), Brussels, Belgium
|
|
IBMM-DBM, Université Libre,
Brussels, Belgium
|
|
DBBM-IOC, Fiocruz, Rio de Janeiro, Brazil
|
|
The Genome Mine, Base4 Bioinformatics,
Canada
|
|
CBI EMBnet Node, University of
Beijing, China
|
|
CSC, Otaniemi, Espoo, Finland
|
|
INFOBIOGEN, Villejuif, France
|
|
Institut Pasteur, Paris, France
|
|
DKFZ,
Heidelberg, Germany
|
|
EMBL, Heidelberg, Germany www.embl-heidelberg.de:80/srs5/
|
|
GBF, Braunschweig, Germany
|
|
INCBI EMBnet Node, Dublin, Ireland
|
|
Weizmann Institute BCD, Rehovot,
Israel
|
|
IVR, Kyoto University, Japan
|
|
Biotek EMBnet Node, Oslo, Norway
|
|
BIC, National University Hospital,
Singapore
|
|
Biomedical Centre (BMC), Uppsala, Sweden
|
|
ExPASy, Geneva, Switzerland
|
|
CAOS/CAMM Center, Nijmegen, The
Netherlands
|
|
Adlib, CAB International,
Wallingford, UK
|
|
EMBL-EBI, Hinxton, Cambridge, UK srs.ebi.ac.uk:5000/srs5/
|
|
HGMP-RC, Hinxton, Cambridge, UK
|
|
MBDC Oxford, Oxford University, UK
|
|
SEQNET EMBnet Node, Daresbury, UK
|
|
Sanger Centre, Hinxton, Cambridge, UK
|
|
IUBio, Indiana University, USA
|
SRSWWW
(http://srs.ebi.ac.uk:5000/srs5/man/srsman.html)
Introduction
SRSWWW is a World
Wide Web interface to the Sequence Retrieval System (SRS). Compared with other
SRS interfaces SRSWWW has more users because of the widespread use of web
browsers, its easiness to handle and its user friendly interface. It supports
HTML3 and lesser versions of HTML. Since HTTP is stateless, a user ID is
created to keep the state for the whole session.
A SRS session is
started by clicking on the 'Start' button in the SRS home page. The page
appearing next is the 'Select Libraries Page' also referred to as 'Top Page'.
Most pages in the
SRSWWW have six header buttons which contain links to other SRSWWW pages. One
header button is the 'Top Page' which links to the start
page of the SRSWWW. The other buttons point to the 'Query
Form' page, the 'Query Manager' page, the 'View Manager' page, the 'Databanks'
page and the 'Help' page.

|
SRSWWW
|
The Top Page
|
|
The 'Top Page'
is used to select the one or more databank(s) to be
searched. Selecting databank(s) can be done by clicking the check boxes. In
all cases, the databank names are linked to an information page ('Information
about ...' page). It provides some information about the indices of the
respective databank. The data fields of the databank can be browsed directly
(see the 'Browse Index' page).
If you want to
change databases for another search you have to go to the 'Top Page'.
|
|
Continue & Reset
Button
|
The 'Reset'
button can be used to deselect all selections done so far. Each databank can
individually be deselected by clicking again their checkboxes. Clicking the
'Continue' button finishes the 'Top page' and brings up the 'Query Form'
page.
|
|
|
|
|
Grouping the Databanks
|
In the 'Top
Page' all available databanks are collected in groups defined by SRS.
|
|
User defined Databases
|
There is a
possibility to include user defined databanks. For example with the search
engine FASTA or BLAST special databanks can be created. Before such a
databank can be selected it has to be created with a search (for instance with BLAST). An existing sequence can be inserted
in the 'Enter query sequence' field by "cut and paste".
|

|
SRSWWW
|
The Query Form Page
|
|
With the 'Query
Form' page the databank query can be defined. The selected databank(s) is/are
listed at the top of the page. Which fields are listed depends on the 'Show
only fields that selected databanks have in common ' check box from the Top Page and the searchable fields of the databank(s).
|
|
Do Query & Reset
Button
|
The 'Reset'
button can be used to deselect all selections done so far. Each selection can
be deselected individually by clicking again the check box. Clicking on the
'Do Query' button starts the query and brings up the 'Query
Result' page.
|
|
FIELD NAME, QUERY,
INCLUDE IN LIST & RETRIEVE SUBENTRIES columns
|
The 'Field Name'
selector lists all data fields of the selected databank which are linked to
an information page. By clicking on "Info" after selecting a 'Field
Name' one moves to the 'Browse Index' page. The
'Query' column is the input field for the query. By using
the operators "&" (= AND), "!" (= NOT), "|"
(= OR) one can use more than one search expression in
every 'Query' input field. The 'Include fields in output'
column is a selection list for inclusion of the corresponding field in the
query result.
|
|
Additional Menus
|
Some fields can
have additional menus with "greater than" and "greater than or
equal to" and "less than" and "less than or equal
to" symbols (e.g., SeqLength) for their range selection. (e.g.,
seqLength 100 && seqLength < 500).
|
|
Combine Searches with
|
If more than one
data field is to be searched the 'Query' input fields can be combined with
the boolean operators 'AND', 'OR' and 'NOT'. Optionally, a wildcard is
appended to every search string to enhance the possibility to find a
match.
|
|
Chunk Size & Use
view
|
Additional
options are 'Entry list in chunks of' to define the number of entries shown
per page and 'Use view' to select a predefined view with which the query
result is shown (see the 'Create a new view' page
for more information on 'Views'). A new view can be defined before doing a
query. All the 'Views' which are applicable to the selected databanks are
listed in the 'Use view' box and one of them can be selected. Note: only the
'Views' are listed which can be used with the selected databank(s). The
default is 'Names Only' (i.e. without a special 'View').
|

|
SRSWWW
|
Browse Index Page
|
|
From the 'Query
Form' page the 'Browse Index' page is opened by clicking on a 'Field Name'
(e.g., the 'ID' field).
|
|
List Values Button
|
Submits a search
string typed in the open box marked with a wildcard (the wildcard can be used
in the string or deleted). The result of any matches (referred to as
"values") of the search string in the indicated 'Field Name' is
listed in the 'Values in the index of the ... field' page. In this list
'Values' can be selected and with the 'Make Query' button a query can be
started immediately with the selected 'Value(s)'.
|

|
SRSWWW
|
The Query Result Page
|
|
The 'Query
Result' page shows the result of a submitted query. The query string, the
number of "hits" and found entries with check boxes and included
fields (if selected in the 'Query Form' page or in the view definition) are
indicated. The found entries are listed by the searched databank and the
entry ID. Clicking on the entry opens the 'Single
Entry' page. In addition, entries can be selected with the checkboxes.
The 'Reset' button can be used to deselect all selections done so far. With
the 'Link', 'Receive' and 'View' buttons further manipulations can be
performed with the selected entries.
|
|
Link Button
|
With this button
the whole/selected result entries can be checked whether they are linked to
other databanks (see the 'Link Page' for more
information).
|
|
Save Button
|
With the
'Receive' button the selected entries can be downloaded.
|
|
Launch
View Button
|
Launch desired
application for further analysis.
With this button
the result entries can be viewed with predefined 'View' definitions. The
'View' definitions can be selected out of the 'View' box.
|
|
Chunk Hypertext Links
|
The last line
are hypertext links for any additional chunk set(s). These links are only
present if the number of entries found is bigger than the number of the
defined chunk size. The chunk set numbers starts from 1 to "number of
hits" divided by "entry list chunk size" (selected in the
'Query Form' page). The user can jump to any additional chunkset by the
hypertext link. The current chunk set is shown within braces. The left
anglebracket hyperlink moves to the first chunkset whereas the right
anglebracket hyperlink moves to the last chunkset.
|

|
SRSWWW
|
Single Entry Page
|
|
Every entry
found and listed in the 'Query Result' page has a link to the complete entry
in the databank. By clicking onto a entry, the 'Single Entry' page comes up
and shows the complete entry. The complete entry can be saved or printed
(refer to the documentation of your web browser).
|

|
SRSWWW
|
Link Page
|
|
From the 'Query
Result' page one can go to the 'Link' page by pressing the 'Link' button. The
'Link' page is used to find the links between the found entries (the set) and
the selected databanks. The user can choose one of three options to display
the different entries. The 'Link' page lists all the databanks like the top
page. The user should select the databanks which are different from the
databanks selected for the query search and should select one of three
options of the 'Find all entries' field. Clicking on the 'Continue' button
shows the 'Query Result' page. A link can also be done in the 'Query Manager' page.
(For more
information about links
refer to the SRS manual)
|
|
Find all entries:
|
|
|
in the selected
DATABANKS which are linked to the current set
|
the result are
entries out of the selected databanks, which are linked with the set.
|
|
in the current SET
which are linked to all selected databanks
|
the result are
entries out of the set, which have links to the selected databanks.
|
|
in the current set
which are not linked to any of the selected databanks
|
the result are
entries out of the set, which contain NO links to the selected databanks.
|

|
SRSWWW
|
Query Manager Page
|
|
The 'Query
Manager' has two functions: one is to store the queries done so far. The
queries are listed in a table. Every query is listed with a check box, the
query name in the form 'Qn' (e.g., Q1, Q2, ..), a type (e.g., 'query' ,
'link' ), the total number of entries found, the library name(s), the number
of entries for each library, the query expression (SRS query syntax)
and a comment. The user may add own commentary to the query. Users working in
HTML3 mode get a table with all these descriptors. In versions below HTML3
the same information is available in different style and in different
order.
The second
function of the 'Query Manager' is to make further queries and links.
There are
three functions to control the queries and three functions to do further
queries and links.
|
|
Control Functions
|
|
|
Save
|
With the 'Save'
function the selected queries can be downloaded.
|
|
Delete
|
The 'Delete'
function deletes selected queries.
|
|
View
|
The 'View'
function can be used to inspect a single query again or with a different
'View' (choose a 'View' from the 'Using the view' field).
|
|
Query/Link Functions
|
|
Link
|
The 'Link'
function is exactly the same as in the 'Query Result' page, see detailed
information in the chapter 'The Query Result Page'.
|
|
Combine
|
With this
function it is possible to combine one or more queries with the logical
operators AND, OR or NOT.
|
|
Expression
|
With this
function the user can directly enter a query like "(Q1 & Q2) ! (Q3 |
Q4) PDB" (i.e. query 1 AND query 2 but NOT query 3 OR query 4 linked to
the PDB database). In contrast to the 'Combine' function it allows to build
more complex queries.
For more
information in using logical
operators and link operators
see the SRS manual.
|
|
Result Display Options
|
|
The 'Query
Manager' offers two options to manipulate the 'Query Result' page:
|
|
Entry list in
chunks of... & Using the view...
|
The options
'Entry list in chunks of ' and 'Using the view' are already described in the
chapter about the 'Query Form' page.
|
The queries done
so far will be lost after closing the SRS session. However, it is quite useful
sometimes (especially in the case of complex query runs) to store a useful
query set in a file (referred to as "history") though it can be
loaded and used again in a new SRS session. This task is
accomplished by activating one or more checkboxes on the bottom 'Save in
histories queries of type' line.

|
SRSWWW
|
View Manager Page
|
|
The extraction
of information from the databank(s) is done with a query created in the
'Query Form' page. In the 'View Manager' you can define how to look at the
query results. You can use predefined 'Views' or create your own 'Views'. You
can apply them on new created queries or already existing queries. A set of
'Views' can be defined before any query has been performed. But of course, a
'View' can only be used after a query has been created.
But the
'View Manager' can do a lot more than only to provide a special service on
query sets as described above. The 'View Manager' works independently from
the 'Query Form' routine. Thus, you can start complex queries within the
'View Manager' to investigate different relations between databanks and/or
query sets.
The following
options are offered in the 'View Manager' page: 'Create a new view', 'Edit
selected view' or 'Delete selected view'. The 'View Manager' lists all the
'Views' defined so far in a table with View Name, Format, Root Libs, Root
Fields, Leaf Libs and Leaf Fields. After doing a query on the selected
database(s) the user gets a set of entries as a result. 'Views' can help to
recognize whether these entries may have e.g., links to other databases.
'Views' can be configured to see the fields of interest from the set and from
the linked database entries. There exist already predefined 'Views' which can
be used directly or modified for your own purpose.
|
|
independent View
Manager
|
Click on the
link 'independent View Editor' with the middle or right mouse button; choose
'Open in New Window' in the appearing context menu. A new browser window is
opened. By this way one can define several 'Views' at a time or edit a 'View'
in one window and use it at once in the 'Query Result' page in the second
window.
|

|
SRSWWW
|
Create A New View Page
|
|
In the 'Create A
New View Page' the available databanks are listed in two columns. The
databanks in the first column are called root databanks. The databanks
in the second column are called leaf databanks. The root databanks
refer to the databanks used for a particular query search. The leaf databank
refer to the databanks which will be linked by the result set of the query
search done with the root databanks. For example the user can define a view
with SwissProt as root databank and Prosite as leaf databank. In the 'View
Select' page the fields which have to be displayed from both databanks have
to be selected. Choose this as a current view and make a query on SwissProt
(which is the root databank). The result will be SwissProt entries and for
entries with a link to Prosite the Prosite entries.
|
|
Name of new view
|
In the 'Name of
view' field you can assign a name to the new 'View'. Leaving this field blank
a 'View' name is created automatically, prefixed by the first root databank
name and a view number at the end (e.g., Swissprot_view1, Prosite_view5
etc.).
|
|
Display view as table
|
With selecting
the 'Display view as table' checkbox the result is displayed in a table form
instead of a list form.
|
|
Print short field
names in header
|
With this
checkbox it is possible to place the short field names as the header of the
table columns.
|
|
Show only
fields common to selected root databanks
|
This checkbox is
selected by default. So only common fields of the root databanks are
listed.
|
|
Continue & Reset
Button
|
The 'Reset'
button can be used to deselect all selections done so far. Each field and
checkbox can individually be deselected by clicking again. Clicking on the
'Continue' button finishes the 'Create new view' page and brings up the 'View Field Select' page.
|

|
SRSWWW
|
Edit Existing View Page
|
|
In this page a
predefined 'View' can be edited. The 'Edit existing view' page offers the
same editing options as the 'Create a new view' page.
|

|
SRSWWW
|
View Field Select Page
|
|
Lists the fields
of all selected root and leaf databanks. Fields can be selected by their
checkboxes and are displayed in the query. The display format of the sequence
field can be selected by the format menu selection (e.g., FASTA, PIR, EMBL,
Protein Chart etc.).
The leaf
databank selection has additional options:
|
|
Use query instead link
|
A query string
can be inserted to search for special relations between the root and leaf
databank.
|
|
Use view to display
entries
|
The result of
the 'View' can be shown by another 'View'.
|
|
Display only number of
linked entries
|
Displays only
the number of links which exist, not the links itself.
|
|
To submit the
view one of the four options ('Top page', 'Query form', 'Query manager' or
'View manager') must be clicked.
|
Databanks Page
The 'Databanks'
page lists all available databases in a table form. Every databank is listed as
a hypertext link and shown with the release version number, the number of
entries, the indexing date, the group (e.g., HomolSearch, Sequence etc.) and an
availability flag. This flag indicates whether the respective databank is
available (ok) or not for searches.
Help Page
Most of the
information about the databanks and their fields description are available
through hypertext links. Help is provided wherever possible in a context
sensitive way. In addition to providing information, the data fields can be
browsed directly (see the 'Browse Index' page).
Principles of sequence comparison
A common problem in
molecular biology is to compare one sequence to another sequence or to a set of
sequences. Previously we have searched databases for text information and
retrieved entries. Sequence comparison, however, is a less trivial problem and
in order to understand it a few theorical considerations are necessary.
Here we will consider
four kinds of sequence comparison:
-
Homology
-
Pattern
-
Multiple sequence alignment
-
Profile
1. Homology
FASTA and BLAST are
programs frequently used to search sequence databases for homology to a search
sequence. Programs of this kind answers questions like : Is my sequence similar
to anything in the database? Did I
finally clone the gene that I’ve been trying to clone for the last three years?
I seem to have identified a new protein, exactly what is the relationship of
this protein to proteins that have been described previously?
One principle to be kept
in mind that sequences are often searched as proteins although they are
determined as DNA sequence. If you have a DNA sequence you may find it natural
to search a DNA sequence database with this sequence. However, often it is more
sensible to use a protein sequence as search sequence. That is, if you think
your DNA sequence encodes a protein first you should try and identify all the
possible translation products of the DNA. Then use the protein sequences to
search a protein database (or a DNA
database where are possible reading frames are examined, a ”translated DNA
database”). Therefore, a common principle is that sequences are determined
as DNA but compared as proteins. The reasons for this are mainly:
-
The genetic code is degenerate. Consider for instance the two codons UCA
and AGU. Both encode serine but are completely unrelated with respect to their
sequence. Therefore, a sequence homology may be significant at
the protein sequence level, but not detectable at the DNA level.
-
All amino acid substitutions do not occur with the same frequency as
further described below (substitution matrices). This is a useful principle
when comparing proteins and there is no equivalent principle for nucleic acid
sequences.
It is important to search
nucleotide databases with proteins as query sequences because protein databases
are not updated quickly enough. Furthermore, DNA databases contain potential
protein coding regions that have not yet been identified. The only drawback
with searching DNA databases with a protein sequence as query is that it is
more time-consuming as all six reading frames have to be examined. TFASTA
and tblastn are programs that do this sort of analysis. An even more time-consuming type of search
is one where a DNA is used as query. All the six possible reading frames are
used as queries to search the six reading frames of a DNA database (tblastx
is one example of such a program, see below)
To understand how
programs like FASTA and BLAST work let’s
compare two related amino acid sequences:
seq 1 A L Q L I W G T S I R D K W P
G D L
seq 2 A L Q M I W A G G T S I R D P
G D L
We can represent them
graphically like this:
SEQ 2
A
*
L * * *
Q *
M
I * *
W *
A
*
G *
*
G *
*
T *
S *
I * *
R *
D *
*
P *
G *
*
D *
*
L * * *
A L Q L I W G T S I R D K W P G
D L SEQ 1
All positions of amino
acid identity are labeled by a ”*” (The GCG program COMPARE in combination with
DOTPLOT produces such a plot). Visual inspection reveals a number of diagonals.
Many computer programs designed to compare sequences identify such diagonals
and tries to combine them into a more extensive alignment. In our example:
seq 1 A L Q L I W - - G T S I R D K
W P G D L
| | | | |
| | | | | | | | | |
seq 2 A L Q M I W A G G T S I R D -
- P G D L
Thus, for a maximal
alignment we have created gaps in each sequence.
A program like FASTA
searches a database for sequence homology using a query sequence using the
principle outlined above. FASTA uses parameters such as:
ktup or wordsize
-
Length of initial peptide match, default is 2, i.e. the program starts
identifying a diagonal by extending a dipeptide match. You may prefer to use
ktup=1 if you want a more sensitive search. (This type of search takes a longer
time, though!)
Gap penalty
Gaps are rarely allowed
in evolution and a database search
program is slow when you instruct it to examine every possible diagonal . For
these reasons a gap penalty is assigned to gaps. These two parameters are
frequently used in homology programs (such as the GCG programs GAP, BESTFIT,
FASTA)
-
Gap weight (Number of gaps)
-
Gap length weight (Total length of gaps)
Substitution matrices
What is taken into accont
in the figure above is only whether there is an exact match or not.
However, we also have to into consideration
whether some matches or mismatches are better than others. To decide on this we
can assign a score to each amino acid
pair with a high score to matches or mismatches that frequently occur in
proteins and a low score to mismatches that seldom occur. To do this we make
use of a table (substitution matrix) that
looks something like this:
Substitution matrix (PAM250)
A R
N D C Q E
G H I L K
M F P S T W Y
V B Z X *
A 2 -2 0 0
-2 0
0 1 -1 -1 -2 -1 -1 -3 1
1 1 -6 -3 0
0 0 0 -8
R 6 0 -1 -4
1 -1 -3 2 -2 -3 3 0
-4 0
0 -1 2 -4 -2 -1 0 -1 -8
N 2 2 -4
1 1 0 2 -2 -3 1 -2 -3
0 1 0 -4 -2 -2 2 1 0
-8
D 4
-5 2
3 1 1 -2 -4 0 -3 -6 -1 0 0
-7 -4 -2 3 3 -1 -8
C 12 -5 -5 -3 -3 -2 -6 -5 -5 -4 -3 0 -2 -8
0 -2 -4 -5 -3 -8
Q
4 2 -1 3 -2 -2 1 -1 -5 0 -1 -1 -5 -4 -2 1 3 -1 -8
E
4 0 1 -2 -3 0 -2 -5 -1 0 0
-7 -4 -2 3 3 -1 -8
G
5 -2 -3 -4 -2 -3 -5 0 1 0
-7 -5 -1 0 0 -1 -8
H 6 -2 -2
0 -2 -2 0 -1 -1 -3 0 -2
1 2 -1 -8
I 5
2 -2 2 1 -2 -1 0 -5 -1 4 -2 -2 -1 -8
L 6 -3 4 2 -3 -3 -2 -2 -1 2 -3 -3 -1 -8
K 5 0 -5 -1 0 0 -3 -4 -2
1 0 -1 -8
M 6 0 -2 -2 -1 -4 -2 2 -2 -2
-1 -8
F 9 -5 -3 -3 0 7
-1 -4 -5 -2 -8
P 6 1 0 -6 -5 -1 -1 0 -1 -8
S 2 1 -2 -3 -1
0 0 0 -8
T 3 -5 -3 0 0
-1 0 -8
W 17 0 -6 -5 -6 -4 -8
Y 10 -2 -3 -4
-2 -8
V 4 -2 -2
-1 -8
B
3 2 -1 -8
Z 3 -1 -8
X
-1 -8
*
1
The table was constructed
from an alignment of proteins, such as cytochrome c from a large number of
organisms. From each amino acid position in this alignment a probability is
calculated for all the possible amino acid replacements, for instance the
probability that serine replaces alanine. In nature that replacement is the
result of one or more mutations at the DNA level. The table of probabilities
was normalized (such that for every 100 amino acids an average of 1 mutation
was accepted --> accepted point mutation = PAM100). The PAM100 table was
calculated by comparing closely related sequences To estimate more distant
relationships a PAM250 was created.) Essentials : Postive values reflect
conservative changes, negative = changes that are unlikey to occur. W and C are
amino acids that are particularly conserved.
PAM tables have been
widely used. Recently however, BLOSUM (Blocks substitution matrices) tables
have become more popular as they often seem to give a somewhat better result.
BLOSUM tables were generated by comparing short conserved regions (BLOCKS) of
proteins. BLOSUM matrices seem to favour hydrophilic matches and aromatic
mismatches.
Low complexity masking
Filter out short repeats
For these parameters see
BLAST below.
BLAST
Speed is increased
compared to FASTA :
-
Wordsize = 3-4
-
No gaps are allowed.
BLAST is therefore less
sensitive than FASTA and less suitable for comparing nucleotide sequences.
BLAST programs
blastp
compares an amino acid query
sequence against a
protein sequence database;
blastn
compares a nucleotide query sequence
against a
nucleotide sequence database;
blastx
compares the six-frame
conceptual translation
products of
a nucleotide query
sequence (both
strands) against a protein
sequence database;
tblastn
compares a protein
query sequence against
a
nucleotide sequence database dynamically
translated in
all six reading
frames (both
strands).
tblastx
compares the six-frame translations of
a nucleo-
tide query sequence against the
six-frame transla-
tions of a nucleotide sequence
database.
Some BLAST parameters:
EXPECT
The statistical
significance threshold for reporting matches against database sequences; the
default value is 10, such that 10 matches are expected to be foundmerely by
chance, according to the stochastic model of Karlin and Altschul (1990). If the statistical significance ascribed to
a match is greater than the EXPECT threshold, the match will not be reported.
Lower EXPECT thresholds are more stringent, leadingto fewer chance matches
being reported. Fractional values are
acceptable. (See parameter E in the
BLAST Manual).
CUTOFF
Cutoff score for
reporting high-scoring segment pairs.The default value is calculated from the
EXPECT value(see above). HSPs are
reported for a database sequenceonly if the statistical significance ascribed
to themis at least as high as would be ascribed to a loneHSP having a score
equal to the CUTOFF value. Higher
CUTOFF values are more stringent, leading to fewerchance matches being
reported. (See parameter S inthe BLAST
Manual). Typically, significance
thresholdscan be more intuitively managed using EXPECT.
MATRIX
Specify an
alternate scoring matrix for BLASTP, BLASTX,
TBLASTN and TBLASTX. The default
matrix is BLOSUM62 (Henikoff & Henikoff, 1992). The valid alternative choices include: PAM40, PAM120, PAM250 and IDENTITY. No alternate scoring matrices
are available for BLASTN; specifying the MATRIX directive in BLASTN requests
returns an error response.
STRAND
Restrict a TBLASTN
search to just the top or bottom strand of the database sequences; or restrict
a BLASTN, BLASTX or TBLASTX search to just reading frames on the top or bottom
strand of the query sequence.
FILTER
Mask off segments
of the query sequence that have low compositional complexity, as determined by
the
SEG program of
Wootton & Federhen (Computers and Chemistry, 1993), or segments consisting
of
short-periodicity
internal repeats, as determined by the XNU program of Claverie & States
(Computers
and Chemistry,
1993), or, for BLASTN, by the DUST
program of Tatusov and Lipman (in preparation).
Filtering can
eliminate statistically significant but
biologically uninteresting reports from the blast
output (e.g., hits
against common acidic-, basic- or proline-rich regions), leaving the more
biologically
interesting
regions of the query sequence available for specific matching against database
sequences.
Low complexity
sequence found by a filter program is substituted using the letter
"N" in nucleotide sequence
(e.g., "NNNNNNNNNNNNN") and the letter "X" in
protein sequences (e.g.,
"XXXXXXXXX"). Users may turn
off filtering by using the
"Filter" option on the "Advanced options for the BLAST server" page.
Filtering is only
applied to the query sequence (or its translation products), not to database
sequences. Default filtering is DUST
for BLASTN, SEG for other programs.
It is not unusual
for nothing at all to be masked by SEG, XNU, or both, when applied to sequences
in SWISS-PROT, so filtering should not be expected to always yield an
effect. Furthermore, in some cases,
sequences are masked in their entirety, indicating that the statistical
significance of any matches reported against the unfiltered query sequence
should be suspect.
Example of BLAST output:
BLASTP 1.4.6MP [13-Jun-94] [Build
13:58:36 Sep 22 1994]
Reference:
Altschul, Stephen F., Warren Gish, Webb Miller, Eugene W. Myers,
and David J. Lipman (1990). Basic local alignment search tool. J. Mol. Biol.
215:403-10.
Query =
pir|A01243|DXCH 232 Gene X
protein - Chicken (fragment)
(232 letters)
Database:
SWISS-PROT Release 29.0
38,303 sequences; 13,464,008 total letters.
Searching..................................................done
Observed Numbers of Database Sequences Satisfying
Various EXPECTation Thresholds (E parameter values)
Histogram units:
= 31 Sequences : less than 31
sequences
EXPECTation Threshold
(E parameter)
|
V Observed Counts--
10000 4863 1861
|============================================================
6310 3002 782
|=========================
3980 2220 812
|==========================
2510 1408 303 |=========
1580 1105 393
|============
1000 712 179 |=====
631 533 161 |=====
398 372 80 |==
251 292 73 |==
158 219 50 |=
100 169 32 |=
63.1 137 18 |:
39.8 119 9 |:
25.1 110 6 |:
15.8 104 9 |:
Expect = 10.0, Observed = 95
<<<<<<<<<<<<<<<<
10.0 95 4 |:
6.31 91 3 |:
3.98 88 1 |:
2.51 87 3 |:
1.58 84 0 |
1.00 84 2 |:
Smallest
Sum
High Probability
Sequences producing High-scoring
Segment Pairs: Score P(N)
N
sp|P01013|OVAX_CHICK GENE X PROTEIN (OVALBUMIN-RELATED) (... 1191
7.7e-160 1
sp|P01014|OVAY_CHICK GENE Y
PROTEIN (OVALBUMIN-RELATED).
949 7.0e-127 1
sp|P01012|OVAL_CHICK OVALBUMIN
(PLAKALBUMIN). 645 3.4e-100
2
sp|P19104|OVAL_COTJA
OVALBUMIN. 626 1.2e-96
2
sp|P05619|ILEU_HORSE LEUKOCYTE
ELASTASE INHIBITOR (LEI). 216 3.7e-71
3
sp|P80229|ILEU_PIG LEUKOCYTE ELASTASE INHIBITOR (LEI)
(... 325 4.0e-71 2
sp|P29508|SCCA_HUMAN SQUAMOUS
CELL CARCINOMA ANTIGEN (SCC...
439 3.5e-70 2
sp|P30740|ILEU_HUMAN LEUKOCYTE
ELASTASE INHIBITOR (LEI) (... 211 1.3e-66
3
sp|P05120|PAI2_HUMAN PLASMINOGEN
ACTIVATOR INHIBITOR-2, P... 176 1.8e-65
4
sp|P35237|PTI_HUMAN PLACENTAL THROMBIN INHIBITOR. 473 1.3e-61 1
sp|P29524|PAI2_RAT PLASMINOGEN ACTIVATOR INHIBITOR-2,
T... 183 9.4e-61 4
sp|P12388|PAI2_MOUSE PLASMINOGEN
ACTIVATOR INHIBITOR-2, M... 179 1.8e-60
4
sp|P36952|MASP_HUMAN MASPIN
PRECURSOR.
198 2.6e-58 4
sp|P32261|ANT3_MOUSE
ANTITHROMBIN-III PRECURSOR (ATIII).
142 4.0e-48 5
sp|P01008|ANT3_HUMAN
ANTITHROMBIN-III PRECURSOR (ATIII).
122 7.5e-48 5
WARNING: Descriptions of 80 database sequences were
not reported due to the
limiting value of parameter V = 15.
... alignments with the top 8 database sequences deleted ...
sp|P05120|PAI2_HUMAN PLASMINOGEN ACTIVATOR INHIBITOR-2, PLACENTAL
(PAI-2)
(MONOCYTE ARG- SERPIN).
Length = 415
Score = 176 (80.2 bits), Expect = 1.8e-65, Sum P(4) = 1.8e-65
Identities = 38/89 (42%), Positives = 50/89 (56%)
Query: 1 QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNN
60
+I +LL S D DT
+VLVNA+YFKG WKT F + PF V
+ PVQMM +
Sbjct: 180 KIPNLLPEGSVDGDTRMVLVNAVYFKGKWKTPFEKKLNGLYPFRVNSAQRTPVQMMYLRE
239
Query: 61 SFNVATLPAEKMKILELPFASGDLSMLVL 89
N+ + K +ILELP+A L+L
Sbjct: 240 KLNIGYIEDLKAQILELPYAGDVSMFLLL 268
Score = 165 (75.2 bits), Expect = 1.8e-65, Sum P(4) = 1.8e-65
Identities = 33/78 (42%), Positives = 47/78 (60%)
Query: 155 ANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRADHPFLFL
214
AN +G+S L
+S+ H A ++++E+G E A TG +
+ QF ADHPFLFL
Sbjct: 338 ANFSGMSERNDLFLSEVFHQAMVDVNEEGTEAAAGTGGVMTGRTGHGGPQFVADHPFLFL
397
Query: 215 IKHNPTNTIVYFGRYWSP 232
I H T I++FGR+ SP
Sbjct: 398 IMHKITKCILFFGRFCSP 415
Score = 144 (65.6 bits), Expect = 1.8e-65, Sum P(4) = 1.8e-65
Identities = 26/62 (41%), Positives = 41/62 (66%)
Query: 90 LPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTSVLMALGMTD
149
+ D + LE
+E I ++KL +WT+ + M + V+VY+PQ K+EE Y L S+L ++GM D
Sbjct: 272 IADVSTGLELLESEITYDKLNKWTSKDKMAEDEVEVYIPQFKLEEHYELRSILRSMGMED
331
Query: 150 LF 151
F
Sbjct: 332 AF 333
Score = 61 (27.8 bits), Expect = 1.8e-65, Sum P(4) = 1.8e-65
Identities = 10/17 (58%), Positives = 16/17 (94%)
Query: 81 SGDLSMLVLLPDEVSDL 97
+GD+SM +LLPDE++D+
Sbjct: 259 AGDVSMFLLLPDEIADV 275
WARNING: HSPs involving 86
database sequences were not reported due to the
limiting value of parameter B = 9.
FASTA
Two major parts of the
program :
-
Search in the database, the best 100-200 database sequences that best
matches the query are saved.
-
These sequences are carefully examined and the best possible alignments
with the query sequence is determined. This step yields an optimized score,
"opt". (FASTA optimizes only in the final step. However, it is
possible to look for optimal alignment of a query with every sequence in the
database. This is referred to as a Smith and Waterman search.)
If you use FASTA you will
most likely find it slower than BLAST.
An advantage with GCG FASTA is that you can search a subset of the EMBL
or Swissprot databases and in this way reduce the time considerably.
TFASTA is a variation of
the FASTA program that searches a nt database with protein query. Therefore,
TFASTA takes six times longer time to run than FASTA.
FASTA output
Histogram Key:
Each histogram symbol represents
128 search set sequences
Each inset symbol represents 1
search set sequences
Score Init1 Initn
(‑) (+)
< 2 5 5:=
4 0 0:
6 3 3:=
8 22 22:=
10 72 72:=
12 341 341:===
14 394 394:====
16 1304 1304:===========
18 1525 1525:============
20 4001
4001:================================
22 5729
5729:=============================================
24 6806
6806:======================================================
26 7626
7626:============================================================
28 6416
6416:===================================================
30 4848
4592:====================================‑‑
32 3710
3281:==========================‑‑‑
34 2457
2081:=================‑‑‑
36 1740 1441:============‑‑
38 1240 947:========‑‑
40 789 638:=====‑‑
42 517 483:====‑
44 292 496:===+
46 190 437:==++
48 124 387:=+++
50 81 316:=++
52 53 249:=+
54 39 182:=+
56 15 153:=+
58 13 119:=
60 8 91:=
62 20 74:=
64 2 36:=
66 4 33:=
68 1 21:=
70 1 15:=
72 0 20:+
74 0 10:+ :++++++++++
76 0 17:+ :+++++++++++++++++
78 1 5:= :=++++
80 0 2:+ :++
82 0 2:+ :++
84 0 3:+ :+++
86 0 4:+ :++++
88 0 1:+ :+
90 0 4:+ :++++
92 0 2:+ :++
94 0 1:+ :+
96 0 0: :
98 0 2:+ :++
100 0 0: :
>100 19 19:= :===================
The best scores are: init1 initn opt..
swissprotold:sr54_mycmy Q01442
mycoplasma mycoides. sign...2044
2044 2044
swnew:sr54_mycge P47294 mycoplasma
genitalium. signal re... 697 773 1038
swissprotold:sr54_bacsu P37105
bacillus subtilis. signal... 477
705 1023
swissprotold:sr54_ecoli P07019
escherichia coli. signal ... 343
588 892
swissprotold:sr54_haein P44518
haemophilus influenzae. s... 341
584 888
swissprotold:sr5c_arath P37107
arabidopsis thaliana (mou... 470
518 895
swissprotold:sr54_yeast P20424
saccharomyces cerevisiae ... 370
435 593
swissprotold:sr54_canfa P13624
canis familiaris (dog). s... 335
413 641
swissprotold:sr54_mouse P14576 mus
musculus (mouse). sig... 329 407 635
swissprotold:sr54_arath P37106
arabidopsis thaliana (mou... 343
379 607
swissprotold:sr54_schpo P21565
schizosaccharomyces pombe... 351
351 649
swissprotold:ftsy_haein P44870
haemophilus influenzae. c... 153
304 404
swnew:ftsy_mycge P47539 mycoplasma
genitalium. cell divi... 151 283 321
swissprotold:pila_neigo P14929
neisseria gonorrhoeae. pr... 176
257 388
swissprotold:dock_sulso P27414
sulfolobus solfataricus, ... 254
254 458
swissprotold:srpr_yeast P32916
saccharomyces cerevisiae ... 155
224 223
swissprotold:ftsy_ecoli P10121
escherichia coli. cell di... 121
208 407
swissprotold:srpr_human P08240 homo
sapiens (human). sig... 121 155 274
swissprotold:srpr_canfa P06625
canis familiaris (dog). s... 121
121 267
swissprotold:rest_human P30622 homo
sapiens (human). res... 63 98
79
swissprotold:srmb_ecoli P21507
escherichia coli. atp‑dep...
61 98 97
swissprotold:gyrb_mycpn P22447
mycoplasma pneumoniae. dn... 50 93
60
swissprotold:flhf_bacsu Q01960
bacillus subtilis. flagel... 77 92
168
swissprotold:hs7c_caeel P27420
caenorhabditis elegans. h... 48 92
53
swissprotold:gr78_rat P06761 rattus
norvegicus (rat). 78... 49 90
52
swissprotold:gr78_mesau P07823
mesocricetus auratus (gol... 49 90
52
swissprotold:gr78_human P11021 homo
sapiens (human). 78 ... 49 90
52
swissprotold:gr78_mouse P20029 mus
musculus (mouse). 78 ... 49 90
52
swissprotold:trj2_ecoli P05837
escherichia coli. traj pr... 47 88
54
swissprotold:rnha_human Q08211 homo
sapiens (human). atp... 49 86
59
swissprotold:rrpl_hrsva P28887
human respiratory syncyti... 49 85
57
swissprotold:ycf1_tobac P12222
nicotiana tabacum (common... 57 85
77
swissprotold:dnak_mycpa Q00488
mycobacterium paratubercu... 53 85
75
swissprotold:vl2_hpv49 P36762 human
papillomavirus type ... 37 84
55
swissprotold:dnak_mycle P19993
mycobacterium leprae. dna... 53 83
80
swissprotold:cse1_yeast P33307
saccharomyces cerevisiae ... 66 83
69
swissprotold:msap_plafw P04933
plasmodium falciparum (is... 45 82
77
swissprotold:pr16_yeast P15938
saccharomyces cerevisiae ... 45 81
56
swissprotold:utro_human P46939 homo
sapiens (human). utr... 41 80
90
swissprotold:kar3_yeast P17119
saccharomyces cerevisiae ... 43 79
88
ID SR54_MYCMY STANDARD; PRT; 447 AA.
AC Q01442;
DT 01‑JUL‑1993 (REL.
26, CREATED)
DT 01‑JUL‑1993 (REL.
26, LAST SEQUENCE UPDATE)
DT 01‑FEB‑1995 (REL.
31, LAST ANNOTATION UPDATE)
DE SIGNAL RECOGNITION PARTICLE
PROTEIN (FIFTY‑FOUR HOMOLOG). . . .
SCORES Init1: 2044 Initn:
2044 Opt: 2044
100.0% identity in 447 aa
overlap
10 20 30 40 50 60
sr54_m MGFGDFLSKRMQKSIEKNMKNSTLNEENIKETLKEIRLSLLEADVNIEAAKEIINNVKQK
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
sr54_m MGFGDFLSKRMQKSIEKNMKNSTLNEENIKETLKEIRLSLLEADVNIEAAKEIINNVKQK
10 20 30 40 50 60
70 80 90 100 110 120
sr54_m ALGGYISEGASAHQQMIKIVHEELVNILGKENAPLDINKKPSVVMMVGLQGSGKTTTANK
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
sr54_m ALGGYISEGASAHQQMIKIVHEELVNILGKENAPLDINKKPSVVMMVGLQGSGKTTTANK
70 80 90 100 110 120
130 140 150 160 170 180
sr54_m LAYLLNKKNKKKVLLVGLDIYRPGAIEQLVQLGQKTNTQVFEKGKQDPVKTAEQALEYAK
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
sr54_m LAYLLNKKNKKKVLLVGLDIYRPGAIEQLVQLGQKTNTQVFEKGKQDPVKTAEQALEYAK
130 140 150 160 170 180
190 200 210 220 230 240
sr54_m ENNFDVVILDTAGRLQVDQVLMKELDNLKKKTSPNEILLVVDGMSGQEIINVTNEFNDKL
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
sr54_m ENNFDVVILDTAGRLQVDQVLMKELDNLKKKTSPNEILLVVDGMSGQEIINVTNEFNDKL
190 200 210 220 230 240
.......
sr54_mycmy
swnew:sr54_mycge
ID SR54_MYCGE STANDARD; PRT; 446 AA.
AC P47294;
DT 01‑FEB‑1996 (REL.
33, CREATED)
DT 01‑FEB‑1996 (REL.
33, LAST SEQUENCE UPDATE)
DT 01‑FEB‑1996 (REL.
33, LAST ANNOTATION UPDATE)
DE SIGNAL RECOGNITION PARTICLE
PROTEIN (FIFTY‑FOUR HOMOLOG). . . .
SCORES Init1: 697 Initn: 773 Opt: 1038
44.7% identity in 445 aa
overlap
10 20 30 40 50 60
sr54_m MGFGDFLSKRMQKSIEKNMKNSTLNEENIKETLKEIRLSLLEADVNIEAAKEIINNVKQK
| ::||: : ::::|::::
|::|:::: :|||||::||:||||: ::|::|:::::|
sr54_m
MFKAMLSSIVMRTMQKKINAQTITEKDVELVLKEIRIALLDADVNLLVVKNFIKAIRDK
10 20 30 40 50
70 80 90 100 110 120
sr54_m ALGGYISEGASAHQQMIKIVHEELVNILGKENAPLDINKKPSVVMMVGLQGSGKTTTANK
::| |: |:: :: ::|::::||:|||:: |: |: :|:| :|||||||||||| :|
sr54_m TVGQTIEPGQDLQKSLLKTIKTELINILSQPNQELN‑EKRPLKIMMVGLQGSGKTTTCGK
PATTERN MATCHING
Homology searches is used
for finding related sequences and allows you to search with relative long
sequences. Pattern searches is used for finding short sequence patterns in a
single sequence, in a group of sequences or in the databases. For instance, the
pattern "GDSGGP" is typical of serine proteases. When a sequence or
database of sequences is analyzed with respect to this pattern the program only
decides whether there is an exact match or not. Consequently, in pattern
matching one is not concerned with gaps and substitution matrices as in
homology searches.
Patterns may be more
complex than the one shown above for serine proteases. The pattern
corresponding to motif A of the ATP/GTP-binding site, for instance is:
[AG]-x(4)-G-K-[ST]
which is to be interpreted :
One amino acid that can be either A or G, followed by any four amino acids,
followed by G and K and finally one amino acid which is either S or T.
For instance,
AGRCGGKT
GGLGGGKT
AGRSGGKS
AGAAGGKT
are all examples of
sequences that will match the pattern.
(The GCG program
FINDPATTERNS uses this type of search. The search pattern is your own or some
predefined like one in PROSITE. The program MOTIFS specifically search a
protein sequence or set of sequences for the PROSITE motifs. In the GCG program
package the prosite information is in a file "prosite.patterns". This
file may be retrieved by typing at the command line : % fetch prosite.patterns)
Pattern searches are very
useful when you encounter a protein and you have no information about its
structure or biochemical function. By searching with PROSITE motifs you may, it
you are lucky, obtain a clue as to the structure or function of the protein.
Examples of data in
PROSITE:
ID ATP_GTP_A;
PATTERN.
AC PS00017;
DT APR-1990
(CREATED); APR-1990 (DATA UPDATE); NOV-1990 (INFO UPDATE).
DE
ATP/GTP-binding site motif A (P-loop).
PA
[AG]-x(4)-G-K-[ST].
CC
/TAXO-RANGE=ABEPV;
3D 1EFM; 1ETU;
1Q21; 2Q21; 4Q21; 5Q21; 6Q21;
DO PDOC00017;
ID ZINC_FINGER_C2H2; PATTERN.
AC PS00028;
DT APR-1990 (CREATED); JUN-1994 (DATA UPDATE);
NOV-1997 (INFO UPDATE).
DE Zinc finger, C2H2 type, domain.
PA
C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H.
NR /RELEASE=35,69113;
NR /TOTAL=1932(412); /POSITIVE=1891(372);
/UNKNOWN=6(6); /FALSE_POS=35(34);
NR /FALSE_NEG=3; /PARTIAL=1;
CC /TAXO-RANGE=??E?V; /MAX-REPEAT=37;
CC /SITE=1,zinc; /SITE=3,zinc; /SITE=7,zinc;
/SITE=9,zinc;
DR P21192, ACE2_YEAST, T; P07248, ADR1_YEAST,
T; P39413, AEF1_DROME, T;
DR Q00900, AGIE_RAT , T; P41696, AZF1_YEAST, T; Q01954, BASO_HUMAN, T;
DR P41182, BCL6_HUMAN, T; P41183, BCL6_MOUSE,
T; P55201, BR14_HUMAN, T;
DR Q01295, BRC1_DROME, T; Q01296, BRC2_DROME,
T; Q01293, BRC3_DROME, T;
DR P10069, BRLA_EMENI, T; Q01713,
BTEB_RAT , T; Q01522, CF23_DROME, T;
DR P20385, CF2_DROME , T; P19538, CID_DROME ,
T; Q05620, CREA_ASPNG, T;
DR Q01981, CREA_EMENI, T; Q08705, CTCF_CHICK,
T; P49711, CTCF_HUMAN, T;
DR P36197, DEFI_CHICK, T; P23792, DISC_DROME,
T; P26632, EGR1_BRARE, T;
DR P18146, EGR1_HUMAN, T; P08046, EGR1_MOUSE,
T; P08154, EGR1_RAT , T;
DR Q05159, EGR2_BRARE, T; P26633, EGR2_CRILO,
T; P26634, EGR2_DUSTH, T;
DR P11161, EGR2_HUMAN, T; P08152, EGR2_MOUSE,
T; P26635, EGR2_POERE, T;
.
. (Edited
here to save a few Swedish trees...)
.
DR P07939, VSI3_REOVL, F; Q93098, WN8B_HUMAN,
F; P51028, WNT8_BRARE, F;
DR P28026, WNT8_XENLA, F; P20201, Y15K_SSV1 ,
F; P20198, Y5K6_SSV1 , F;
DR P43558, YFE4_YEAST, F; P37127, YFFG_ECOLI,
F; P38890, YH07_YEAST, F;
DR Q09441, YP83_CAEEL, F;
3D 1ARD; 1ARE; 1ARF; 1PAA; 1ZAA; 1AAY; 2GLI;
1SP1; 1SP2; 1NCS; 1ZFD; 1TF3;
3D 2DRP; 1ZNF; 3ZNF; 4ZNF; 1BBO; 5ZNF; 7ZNF;
DO PDOC00028;
Multiple sequence
alignment
The homology search
programs FASTA and BLAST both rely on a basic procedure to compare two
sequences with each other. Multiple sequence alignment programs, on the other
hand, allows you to align and directly compare more than two related sequences. This procedure is a very useful tool if you
want to analyze a family of proteins and for instance to identify the
structural elements that are characteristic of that family. A common program
for multiple sequence analysis is CLUSTALW, or in the GCG package PILEUP.
PILEUP does an alignment
with many of the considerations as for programs like FASTA and BLAST. However,
it is not possible to make such a rigorous comparison of many sequences as when
only two sequences are compared.
Instead, "Pileup" and "Clustalw" performs a pairwise
comparison.
Let's look at the result
of a PILEUP program:
PileUp of:
@../kurs/l5/evol.list
Symbol comparison table: GenRunData:pileuppep.cmp CompCheck: 1254
GapWeight: 3.000
GapLengthWeight: 0.100
evol.msf MSF: 650 Type: P
July 24, 1995 17:08 Check: 7557
..
Name: HS71_HUMAN
Len: 650 Check: 2160
Weight: 1.00
Name: HS71_MOUSE
Len: 650 Check: 3217
Weight: 1.00
Name: HS72_MOUSE
Len: 650 Check:
383 Weight: 1.00
Name: HS70_CHICK
Len: 650 Check: 1880
Weight: 1.00
Name: HS72_YEAST
Len: 650 Check: 3179
Weight: 1.00
Name: HS71_YEAST
Len: 650 Check: 5547
Weight: 1.00
Name: HS74_YEAST
Len: 650 Check: 1191
Weight: 1.00
//
1 50
HS71_HUMAN .MAKAAAIGI DLGTTYSCVG VFQHGKVEII ANDQGNRTTP
SYVAFTDTER
HS71_MOUSE .MAKNTAIGI DLGTTYSCVG VFQHGKVEII ANDQGNRTTP
SYVAFTDTER
HS72_MOUSE MSARGPAIGI DLGTTYSCVG VFQHGKVEII ANDQGNRTTP
SYVAFTDTER
HS70_CHICK MSGKGPAIGI DLGTTYSCVG VFQHGKVEII ANDQGNRTTP
SYVAFTDTER
HS72_YEAST ....SKAVGI DLGTTYSCVA HFSNDRVDII ANDQGNRTTP
SFVGFTDTER
HS71_YEAST ....SKAVGI DLGTTYSCVA HFANDRVDII ANDQGNRTTP
SFVAFTDTER
HS74_YEAST ....SKAVGI DLGTTYSCVA HFANDRVEII ANDQGNRTTP
SYVAFTDTER
---cut to save Swedish trees
---
601 650
HS71_HUMAN ELEQVCNPII SGLYQGAGGP GPGGFGAQ.. ...G.PKGGS
GSGPTIEEVD
HS71_MOUSE ELERVCSPII SGLYQGAGAP GAGGFGAQ.. ...APPKGAS
GSGPTIEEVD
HS72_MOUSE ELERVCNPII SKLYQG.GPG GGG....... .........S
SGGPTIEEVD
HS70_CHICK ELEKLCNPIV TKLYQGAGGA GAG....... .........G
SGGPTIEEVD
HS72_YEAST ELQEVANPIM SKLYQAGGAP EGA...APGG FPGGAPPAPE
AEGPTVEEVD
HS71_YEAST ELQDIANPIM SKLYQAGGAP GGAAGGAPGG FPGGAPPAPE
AEGPTVEEVD
HS74_YEAST ELEGVANPIM SKFY...GAA GGAPGAGPVP GAGAGPTGAP
DNGPTVEEVD
The graphical output file from
PILEUP displays a similarity tree that illustrates the similarity between the
sequences. This gives a good overview about how the sequences are related to
each other. The length of the vertical lines is directly proportional to the
difference between the sequences (or sequence groups). By looking at the figure
below you may estimate that the two sequences 2 and 3 (from left) are the most
similar and they are about twice as similar as 6 and 7 are to each other. 
By looking at this tree it's rather
easy to understand how "Pileup" works. The program creates the tree
by pairing the most similar sequences (or clusters of previously paired
sequences) step by step. It first selects the two sequences that are most
similar (in this case sequence 2 and 3 from the left) and group these two
together. It then takes the next two sequences (or groups) that are most
similar, in this case sequence 6 and 7. The next step is to pair 4 and 5. Now
it pairs the group 4,5 with the group 6,7.
Then sequence 1 to the group 2,3. Finally the two large groups are
joined.
Note that the tree is a similarity
tree. You may be fooled that this is is a phylogenetic tree, but it's not. However, this figure is very informative,
yet simple and easy to interpret.
Profile analysis
Profile analysis is a method of
sequence comparison which is distinct from homology and pattern searches. The starting point is a muliple sequence
alignment. (However, that sequence alignment is not necessarily the result of a
sequence alignment program like PILEUP but could also be an alignment based on
three dimensional structures!) The
alignment is used to create a PROFILE. The profile is a table where we
find for each amino acid position the frequency (somehow normalized) of each of
the 20 amino acids.
Below is the beginning of
a cut version of the output file. The sequence is on the vertical axis, one
aminoacid per line. Each line then contains scores against all aminoacids and,
at the end, for a gap at that position.
Like this:
(Amino acids M - T
have been edited out in this table)
Cons A
B C D E F
G H I K --
V W Y Z Gap
Len ..
M
0 -4 -8 -5 -3
7 -4 -4 8 3
-- 8 -4 -1 -1
24 24
S
6 0 1 -3 0
3 4 -7 7 6
-- 7 0 -7 -1
24 24
A
38 8 8 11 10
-15 25 -3
-2 -1 -- 6 -25
-11 7 24
24
K
-2 10 -16 7 7
-19 -4 6
-6 39 -- -6 11
-17 10 24
24
S
30 23 19 21 17 -21 41
-5 -8 6 -- 0
-14 -21 8
100 100
K
19 13 -9 11 11
-31 9 4
-6 37 -- -1 -18
-28 14 100
100
A
150 20 30
30 30 -50 70 -10
0 0 -- 20 -80
-30 20 100
100
I
5 -11 11 -11 -11
28 -5 -17 75 -11
-- 71 -35 1 -11
100 100
G
70 60 20 70 50
-60 150 -20
-30 -10 --
20 -100 -70 30
100 100
I
0 -20 20 -20 -20
70 -30 -30 150 -20
-- 110 -50 10 -20
100 100
D
30 110 -50
150 100 -100 70
40 -20 30 -- -20 -110
-50 90 100
100
L
-10 -50 -80
-50 -30 120
-50 -20 80
-30 -- 80
50 30 -20 100 100
The GCG program
"Profilemake" creates the profile. The next step is to use the
profile to search a database. The GCG program to use is PROFILESEARCH. In this
way new members of the protein family may be identified.
Profile searching can be effective
for specific structural motifs that cannot be identified by conventional
homology searching. One very illuminating example is the helix-turn-helix (HTH)
motif. A search with a profile created from the HTH motif very effectively
identifies such motifs in the Swissprot protein database. In contrast, using
BLAST or FASTA with a
helix-turn-helix-containing protein as query sequence is useless in
identifying members of this family of proteins.
GCG - an introduction
After you have logged in
to gcg.mednet.gu.se you initiate the GCG package version 9 by the command:
% gcg9
The following text
appears on your screen:
Welcome to the WISCONSIN
PACKAGE
Version 9.0-UNIX,
December 1996
Installed on irix
Copyright 1982-1996, Genetics Computer
Group, Inc. All rights reserved.
Published research assisted by this
software should cite:
Wisconsin Package Version 9.0, Genetics
Computer Group (GCG), Madison, Wisc.
Databases available:
EMBL Release 52.0 (10/97)
SWISS-PROT Release 34.0 ( 9/96)
PROSITE Release 14.0 ( 9/96)
Restriction Enzymes (REBASE) (10/96)
em_new (EMBL since rel.52.0) was
updated 02 March 1998
sw_new (SWISSPROT since rel.34.0) was
updated 31 January 1997
Online help: % genhelp or
http://www.gcg.com/genhelp/
The GCG program package
can be operated in two different ways:
- Command line
- Graphic interface SeqLab
In the command line mode
all programs are operated by commands issued at the command prompt. For
instance to translate the sequence "test.seq" you would have to type
the following at the command prompt :
% translate test.seq
Here we will focus on the
graphic environment provided by SeqLab. To start this interface:
% seqlab &
The symbol '&' is a
UNIX function that means that the program is carried out "in the
background". This means that at any point you may return from a SEQLAB
window to the command line control without closing the SEQLAB.
(For the seqlab to
function it is essential that you have software in your local computer to
emulate X windows and that you have performed a command "setenv DISPLAY
xx.yy.zz.aa:0" where "xx.yy.zz.aa" is the Internet name of your
computer, such as "laban.medkem.gu.se". It is suitable to have this
command a part of your login procedure, i.e in you file .cshrc).
If you want to have a
smaller or larger X window you may instead use
% seqlab -small &
and
% seqlab -large &
respectively.
Click "OK" on
the window in front.
You are now left with the
main SEQLAB window "The SeqLab Main Window on ...".
There is a GCG tutorial
on the SeqLab Interface which is a very good introduction to the environment.
Getting help
In the SEQLAB on-line
help is always available. For instance, in the main window click on
"Help".
In the command line mode
the same help information is available. To access online help organized by
program name , type:
% genhelp
To access online help
organized in sections like the Program Manual, type:
% genmanual
General information about
the Genetics Computer Group is found at
www.gcg.com and a help desk is available at the email address help@gcg.com
Seqlab - short
introduction
There are two main modes
of operation in the SeqLab window:
·
Main list
·
Editor
The default mode is Main
list. Select the Editor mode where you are about to edit a sequence or a
multiple sequence alignment. All programs are carried out in the following way.
Click with the mouse on the sequence (or sequences) that you want to analyze.
Then go to the Functions menu. As you select the program you want to run a
dialog box specific to that program shows up. If you accept the default
settings for the program you simply click on the Run button to start the
program.
To see the result of the
program you must go to Windows - Output
manager . Double click on the result file to see its contents. If the result is
a list file you can add it to your Main list to use it for instance as input to
another program.
If you think you have
started a program but cannot find the result in the Output window you might
find it useful to go the Job manager window (Also under the Windows menu) .
From the list in Job Manager you can for instance see whether a program is
running or if an error occurred during its execution (oops!).
Also under the Windows
menu is the "Database browser" that allows to browse the EMBL and
Swissprot databases in the GCG environment.
Types of sequence files in the
GCG environment
Recommended reading: Users's guide (UNIX)
Wisconsin Package
·
Database sequnces
·
Single
sequence files
·
List files
·
Multiple
sequence format files
Database sequences
Specifying database sequences in the GCG
environment:
Database Valid
names
____________________________________________________________
All DNA sequences em
embl
Bacterial bacterial pro
bact ba
EST est
Fungi fungi fun
Invertebrates in inv
Other Mammalian om
Organelle organelle org
or
Other Vertebrate ov
Patent patent pat
Phage phage ph
Plant pl plant
Primate primate pr
pri
Rodent rodent ro
rod
STS sts
Synthetic synthetic
syn
Unannotated unannotated
un
Viral viral
vir
EMBL‑New,all
divisions
(since latest
release) em_new
Swiss‑Prot swissprot sw
Swiss‑Prot
(since latest
release) swissprotnew
A specific sequence in
these databases may be accessed using the syntax database:entry, where entry
is the AC or ID.
Examples:
em_ba:BA09230
pro:BA09230
pro:U09230
em:U09230
bact:U09230
all refer to the same
sequence where U09230 is the accession code and BA09230 is the ID.
Swissprot entries are
referred to in the same way: sw:ftsy_ecoli and sw:p10121 is the
same sequence (ID and AC respectively). The last five letters of the Swissprot
ID is an abbreviation for the organism.
It is also possible to
use wildcards to specify a set of sequences. Examples:
em:* the
entire EMBL database
ba:* the
bacterial section of EMBL
em:a* all entries of embl beginning with "a".
sw:* all swissprot entries
sw:*ecoli all E. coli
sequences of Swissprot
sw:*human all human sequences of Swissprot.
GCG sequence files
GCG has its own format of
the sequence files. Here is a simple example of a sequence file:
This is just a
test sequence
test.seq
Length: 59 June 22, 1994
19:00 Type: N Check: 9968 ..
1
atctagcacg cgtgcccgat gcatgctgca cgatcgatcg tgtagtcgat
51
gctagctag
At the top is a comment
line. After that comes a GCG specific line containing the name of the sequence,
the length, the date, the type of sequence (N=nucleotide, P=protein) and a
checksum. The purpose of the checksum is to ensure that the sequence is correct
and not corrupted. You can NOT use a standard text editor to edit a GCG
sequence file. You have to use the GCG editor programs to ensure that you get
the proper format and checksum.
List files
An important GCG concept
is "list files". A list file is a textfile containing a list of
sequence files. Here is a simple example:
..
first.seq
second.seq
third.seq
The file begins with two
dots. Anything above this line is treated as a comment. After the two dots
follows a list of sequence files, one per line. It doesn't have to be sequence
files located in your local directory, but it may be sequences located
elsewhere, database entries and even other listfiles. Here is a more
complicated example:
This is an example of a list file
..
myown.seq sequence
file in this directory
/u/medkem/short.seq sequence file in
another directory
inv:ctbr6 database
sequence
@long.listfile another list file
Or this short and simple,
yet very useful list file:
Mammals.listfile - allows me to search ALL
mammal sequences in EMBL
at the same time.
..
hum:*
rod:*
mam:*
List files are very
efficient when working with several sequences at the same time. Several GCG
programs creates list files as output. For instance, FASTA produces a list file
that can be used as input to PILEUP to generate a multiple sequence alignment
from the sequences in the FASATA output file. You may also create your own list
files using a text editor.
In a GCG program you may
refer to all sequences listed in a list file by using the "@"-sign.
As an example look at the following question in stringsearch:
STRINGSEARCH through what sequence(s) (*
GenEMBL *) ? @long.listfile
The answer in this case
means that you want to search inside all sequences listed in the list file
called "long.listfile".
Multiple sequence format
files
Multiple sequences alignments like those created by PILEUP are
stored in a specific format (multiple sequence format, MSF). An example of such
a file is:
..
Name: rmet Len: 1410 Check:
701 Weight: 1.00
Name: mmet Len: 1410 Check: 9020
Weight: 1.00
Name: hmet Len: 1410 Check: 7912
Weight: 1.00
//
1 50
rmet MKAPTALAPG ILLLLLTLAQ RSHGECKEAL VKSEMNVNMK
YQLPNFTAET
mmet MKAPTVLAPG ILVLLLSLVQ RSHGECKEAL VKSEMNVNMK
YQLPNFTAET
hmet MKAPAVLAPG ILVLLFTLVQ RSNGECKEAL AKSEMNVNMK
YQLPNFTAET
51 100
rmet PIHNVVLPGH HIYLGATNYI YVLNDKDLQK VSEFKTGPVV
EHPDCFPCQD
mmet
PIQNVVLHGH HIYLGATNYI YVLNDKDLQK VSEFKTGPVL EHPDCLPCRD
.
.
.
.
1301 1350
rmet APPYPDVNTF DITIYLLQGR RLLQPEYCPD ALYEVMLKCW
HPKAEMRPSF
mmet APPYPDVNTF DITIYLLQGR RLLQPEYCPD ALYEVMLKCW
HPKAEMRPSF
hmet APPYPDVNTF DITVYLLQGR RLLQPEYCPD PYEVMLKCW
HPKAEMRPSF
1351 1400
rmet SELVSRISSI FSTFIGEHYV HVNATYVNVK CVAPYPSLLP
SQDNIDGEAN
mmet SELVSRISSI FSTFIGEHYV HVNATYVNVK CVAPYPSLLP
SDNIDGEGN
hmet SELVSRISAI FSTFIGEHYV HVNATYVNVK CVAPYPSLLS
SEDNADDEVD
1401
rmet T.........
mmet T.........
hmet
TRPASFWETS
Managing the GCG environment - FTP (File transfer protocol). Moving files between
the personal computer and the GCG environment.
Transfer of files from
one computer to another is often done with FTP (file transfer protocol).
Different FTP programs
are available. Examples are CuteFTP (PC) and Fetch (Mac). FTP is also possible
with a web browser like Netscape. When you transfer a sequence from your PC or
Mac to the GCG computer, remember that the sequence should be in a text format.
For instance if you have used Word to produce a document with a sequence, it is
important to save that document as a Text file and not in the standard (binary)
Word format. (Save as ... Select Text
format).
There are two forms of
FTP:
- Anonymous FTP
User anonymous
Password
[your email address]
Example with Netscape: ftp://ftp.gu.se
- Personal FTP
User
[your account username]
Password
[password associated with the username]
Example with Netscape:
ftp://kurs1@gcg.mednet.gu.se (Enter password)
Upload a file with Netscape : Select File -
Upload
FTP is also possible
using command line from a PC or from a UNIX environment.
Some commands (command
line)
! escape to the shell
ascii set ascii transfer type
binary set binary transfer type
bye terminate ftp session and exit
cd change remote working directory
cdup change remote working directory to
parent directory
dir list contents of remote directory
get receive file
image set binary transfer type
ls list contents of remote
directory
idle get (set) idle timer on remote
side
mget get multiple files
mkdir make directory on the remote
machine
mput send multiple files
prompt force interactive prompting on
multiple commands
put send one file
pwd print working directory on remote
machine
quit terminate ftp session and exit
Managing the GCG
environment - Some useful commands in UNIX
What files are there?
In your local directory
you have a number of files, for instance sequences that you use with the GCG
package. To list all files in your directory:
% ls
The option -l gives more
information about the files and the parameter -a lists also those files that
has a dot as the first character.
% ls
-al
A wildcard (*) may be
used to list a number of files with characters in common. For instance the
command below lists all files with the extension "seq":
% ls
*.seq
To copy a file
Sometimes you want to
make a copy of a file. For instance,
you may want to store a result of a GCG program under a specific name so that
the information it is not accidentally written over by a new run of the same
program.
For instance :
% cp test.seq
test2.seq
To check the result of
this operation use the command:
% ls test*
To erase a file
To delete a file use the
command rm, for instance
% rm test2.seq
will delete the file
"test2.seq". Again, use ls to check the result.
To make a new directory
Sooner or later your
working directory gets crowded with files and you may want to organize your
work. You create subdirectories with mkdir. For instance,
% mkdir mydir
will create the directory
"mydir".
To move to this directory
from your home directory type
% cd mydir
To go back to the
directory higher up:
% cd ..
(The directory
"mydir" may be removed by "rmdir mydir")
If you are lost and do
not know in what directory you are the command
% pwd
will present the current
directory.
The command
% cd
will always return you to
your home directory, i.e the directory you are in when you have logged in to
the computer.
If you want to rename a
file or move it to another directory, use the command "mv". For
instance
% mv test.seq test2.seq
will rename the file
"test.seq" to "test2.seq".
% mv test.seq mydir
will move the file
"test.seq" to the subdirectory "mydir"
To look at the contents
of a file
The commands cat
and less will list the contents of a text file. For instance
% cat test.seq
will present the contents
of "test.seq", a nucleotide sequence in GCG format. If you want to
examine a larger file it may be more convenient to use the command less. This
command will list the contents just as cat, but one page at a time. Use
the SPACE key to move to the next page, "u" to move backwards, and
"q" to quit.
% less test.seq
File editing
Different editors are available
in Unix. Examples av text editors that run from a simple text terminal are
"vi", and "emacs". Examples of graphical text editors that
are based on X windows are "jot" and "nedit". The later
editors are very easy to use but require an X windows environment.
Do the following exercise
with the editor "nedit"
% nedit &
In the resulting window
type from the keyboard a short nucleotide sequence. Save the file under a name,
for instance "min.seq". Exit "nedit" (Select File -
Quit) ( You may use % cat min.seq to
see the result of the file editing.)
To make this sequence in
a format used by the GCG programs type:
% reformat min.seq
(You must have issued the
% gcg command before using "Reformat" as it is a GCG program
and not a regular UNIX command. Type % cat
min.seq to see the result of the reformat procedure)
Changing the password
% passwd
Answer the questions:
Old password:
New password:
Retype new password:
Logging out
To end a UNIX session:
% logout
or
% exit
For more information on
UNIX commands see the GCG UNIX manual.
Overview of GCG
programs
COMPARISON
DATABASE_SEARCHING
DISPLAY
EDITING
EVOLUTIONARY_ANALYSIS
FILE_UTILITIES
FRAGMENT_ASSEMBLY
MANIPULATION
MAPPING
MISCELLANEOUS
MULTIPLE_SEQUENCE_ANALYSIS
PATTERN_RECOGNITION
PROTEIN_ANALYSIS
RNA_SECONDARY_STRUCTURE
SEQUENCE_EXCHANGE
TRANSLATION
Database
searching
You can search through the nucleic acid or
protein sequence databases
for sequences similar to your sequence or sequence pattern. You can
also search the documentation in the databases for specific
text
patterns.
Any database sequence and its associated documentation can
be retrieved and written into a personal
file. You can make a data
library of your own sequences.
BLAST LOOKUP FASTA TFASTA WORDSEARCH
SEGMENTS FRAMESEARCH FINDPATTERNS STRINGSEARCH NAMES
FETCH DATASET ToBLAST DBINDEX
COMPARISON
Sequences
can be compared and the comparisons can be visualized
graphically with dot-plots or optimal
alignments.
COMPARE DOTPLOT BESTFIT GAP GAPSHOW FRAMEALIGN
OVERLAP NOOVERLAP
DISPLAY
The Display programs make
publication-quality displays of plasmids,
manuscripts, figures, and sequences.
The output from
any GCG
graphics
program can be made into a
figure and incorporated into a
manuscript that prints out on a PostScript
laser printer. Figure and
Red are the programs we use to publish our
documentation.
PLASMIDMAP FIGURE RED PUBLISH
EDITING
An editor lets you enter or modify protein or nucleic acid sequences.
To make the entry of nucleic acid sequence data easier,
the keys on
the keyboard can be remapped or a digitizer can be used.
SEQED SETKEYS
EVOLUTIONARY_ANALYSIS
The
programs in this
chapter allow you
to investigate the
relationships within a group
of aligned sequences. You can
compute
the pairwise distances between sequences in
an alignment, reconstruct
phylogenetic trees using distance methods, and calculate the degree
of divergence of two protein coding
regions.
DISTANCES GROWTREE NEWDIVERGE DIVERGE
FILE_UTILITIES
These utilities act on text files.
LPRINT REPLACE COMPRESSTEXT ONECASE SHIFTOVER
DETAB CHOPUP CRYPT LISTFILE GETTEXT
COUNT EXAMINE FILECHECK
FRAGMENT_ASSEMBLY
The Fragment Assembly programs let you enter and assemble overlapping
nucleotide sequence fragments
into one continuous
sequence. These
programs are based on the
method of Staden (Nucleic Acids Research
8(16); 3673-3694 (1980)).
GELSTART
GELENTER
GELMERGE
GELASSEMBLE
GELVIEW
GELDISASSEMBLE
MAPPING
Restriction digests can be calculated and displayed by
several
different
programs. A program
is provided to
simulate RNA
fingerprints. You can also identify sites in sequences that would be
ideal for priming.
MAP MAPPLOT MAPSORT PRIME FINGERPRINT
MULTIPLE_SEQUENCE_ANALYSIS
The first four programs in this chapter allow you to create,
edit,
and display multiple sequence
alignments. Another program allows you
to represent the multiple sequence
alignment as a profile, defining
conserved or variable regions. The profile can be used to search the
databases
for similar sequences or in a
pair-wise comparison with
another sequence.
PILEUP LINEUP PRETTY PLOTSIMILARITY
Profile_Analysis
PROFILEMAKE
PROFILESEARCH PROFILESEGMENTS
PROFILEGAP
PATTERN_RECOGNITION
These
programs help you
recognize peptide coding
regions,
terminators, repeats, and other consensus patterns.
Several of the
programs are for the analysis of sequence
composition.
CODONPREFERENCE TESTCODE
FRAMES REPEAT
WINDOW STATPLOT
COMPOSITION TERMINATOR
CONSENSUS FITCONSENSUS
CODONFREQUENCY CORRESPOND
PROTEIN_ANALYSIS
Most GCG programs work on either
protein or nucleotide sequences.
The
programs in this chpater, however, do analyses specific
to
protein sequences. The first two programs identify
sequence motifs
in protein sequences. The next three programs make predictions
about
peptide
isolation. The last
five look at secondary
structure,
hydrophobicity, and antigenicity.
MOTIFS
PROFILESCAN
PEPTIDESORT ISOELECTRIC
PEPTIDEMAP PEPPLOT PEPTIDESTRUCTURE PLOTSTRUCTURE
MOMENT
HELICALWHEEL
RNA_SECONDARY_STRUCTURE
Zuker's MFold program predicts optimal
and suboptimal RNA secondary
structures, which then can be displayed
graphically in any of six
different
ways with the PlotFold
program. Zuker's older
FoldRNA
program predicts optimal RNA secondary structure, which then can be
displayed graphically in any of five different ways. StemLoop looks
for inverted repeats.
MFOLD PLOTFOLD FOLDRNA
SQUIGGLES CIRCLES DOMES
MOUNTAINS
STEMLOOP
SEQUENCE_EXCHANGE
These utilities convert sequences from one
format to another so that
they can be used with different sequence
analysis packages.
REFORMAT FROMSTADEN FROMEMBL FROMGENBANK FROMPIR FROMIG
FROMFASTA TOSTADEN TOPIR TOIG
TOFASTA GETSEQ
SPEW
TRANSLATION
These
programs translate nucleic
acids into proteins and proteins
back into nucleic acids.
TRANSLATE BACKTRANSLATE
EXTRACTPEPTIDE PEPDATA
A selection of the
most important GCG programs
Compare two
sequences
·
COMPARE
·
DOTPLOT
·
BESTFIT
·
GAP
·
FRAMEALIGN
Database
searches
·
BLAST
·
FASTA
·
TFASTA
·
FRAMESEARCH
·
FINDPATTERNS
·
STRINGSEARCH
Mapping
(Restriction and other)
·
MAP
·
MAPPLOT
·
MAPSORT
·
PLASMIDMAP
·
PRIME
Multiple
sequence alignment
·
PILEUP
·
PRETTY
·
PLOTSIMILARITY
Profile
Analysis
·
PROFILEMAKE
·
PROFILESEARCH
·
PROFILESEGMENTS
Pattern
recognition
·
CODONPREFERENCE
·
FRAMES
·
REPEAT
·
COMPOSITION
·
TERMINATOR
·
CONSENSUS
·
CODONFREQUENCY
Protein
analysis
·
MOTIFS
·
PEPTIDESORT
·
PEPTIDEMAP
·
HELICALWHEEL
RNA folding
·
MFOLD
·
PLOTFOLD
·
FOLDRNA
·
SQUIGGLES
·
STEMLOOP
Conversion
between sequence formats
·
REFORMAT
·
FROMEMBL
·
FROMGENBANK
·
FROMFASTA
·
TOSTADEN
·
TOFASTA
Translation
·
TRANSLATE
·
BACKTRANSLATE
Formatting for
publishing
·
PUBLISH
COMPARE
Compare is the first
program of a two-program set that
produces
dot-plots. Compare compares
two sequences and writes a file of the
points where matches of a
certain quality are found. The
points in
the output file
can be plotted with the
DotPlot program.
Dot-plotting is the best method
in the Wisconsin Sequence Analysis
Package(TM) for comparing two
sequences when you suspect that there
could be more than one segment of similarity between the two.
Compare makes a file with the
coordinates of each point where
two
sequences are similar. The
sequences are compared in every
possible
register and a point
is added to the file
wherever some match
criterion for similarity is
met. The match criterion can be met in
two different ways:
The standard way compares two sequences in every
register,
searching for all the
places where a given number of matches
(stringency) occur
within a given range (window).
See Maizel
and Lenk (1981) "Enhanced Graphic Matrix
Analysis of Nucleic
Acid and Protein Sequences"
Proc. Natl. Acad.
Sci. USA 78;
7665-7669 for a description of the matrix analysis of biological
sequences.
The other way to find points of similarity is to search for
short perfect matches of some set length. Short perfect matches
are referred to as words. The
word comparison between
two
sequences is about 1,000 times faster than the window/stringency
match described above,
but it requires that the sequences
contain short perfect matches for
any similarity to be found.
Word comparison is discussed
in detail by Wilbur and Lipman
(1983) "Rapid
Similarity Searches of
Nucleic Acid and Protein
Data Banks" Proc. Natl.
Acad. Sci. USA 80; 726-730. The
authors refer to a word
as a k-tuple. Compare
does a word
comparison if it is run with the command-line option -WORdsize.
You may limit the number of
points that Compare finds
with the
command-line option -LIMit.
DOTPLOT
Dot-plotting is the best way to
see all of the structures in
common
between two sequences or to visualize all of the repeated or inverted
repeated structures in one sequence.
DotPlot is the second part of a
two-part set of programs that generate dot-plots of the points of
similarity between two
sequences. (See Maizel
and Lenk (1981)
"Enhanced Graphic Matrix
Analysis of Nucleic
Acid and Protein
Sequences" Proc. Natl.
Acad. Sci. (USA)
78(12); 7665-7669.)
Compare writes a point file with
the coordinates of the points in
common between two
sequences. DotPlot plots those points
on a
plotter or a graphics terminal.
DotPlot calculates the minimum density
in bases per 100 platen units
along either axis that would allow all of the points to be plotted on
a single page. At high
densities (for example, 1,000 bases per
100
platen units), the dots will not be individually resolved.
You can select any density you want and DotPlot divides the plot into
as many pages as it takes to
plot the whole file. Before you decide
to go ahead, DotPlot tells you how many
pages it would take at your
chosen density, and if you
have chosen your density interactively,
DotPlot gives you a chance to
change your mind. Look at the output
suggestions below for more help on the format.
BESTFIT
BestFit inserts gaps to obtain
the optimal alignment of
the best
region of similarity between two
sequences, and then
displays the
alignment in a format similar to
the output from Gap. The
sequences
can be of very different
lengths and have only a small segment
of
similarity between them. You could take a short RNA sequence, for
example, and run it against a whole mitochondrial genome.
GAP
Gap considers all possible
alignments and gap positions and
creates
the alignment with the largest number of matched bases and the fewest
gaps. You provide a gap creation
penalty and a gap extension penalty
in units of matched bases. In
other words, Gap must make a profit of
gap creation penalty number of
matches for each gap it inserts. If
you choose a gap extension penalty greater than zero, Gap must, in
addition, make a profit for each
gap inserted of the length of the
gap times the gap
extension penalty. Typical values to use as a
point of departure for the
gap creation and gap extension
penalties
are 5.0 and 0.3, respectively, for nucleic acid sequence comparisons,
and 3.0 and 0.1, respectively, for protein sequence comparisons. Gap
uses the alignment method of Needleman and Wunsch (J. Mol. Biol. 48;
443-453 (1970)) that has been shown to
be equivalent to Sellers (see
note below).
FRAMEALIGN
FrameAlign inserts gaps to obtain the optimal local alignment of the
best region of similarity
between a protein sequence and
the codons
in a nucleotide sequence. Because FrameAlign can align the protein
to codons in different reading
frames of the nucleotide sequence, it
can identify sequence similarity
even when the nucleotide sequence
contains reading frame shifts.
In standard sequence
alignment programs, you routinely specify gap
creation and extension penalties. In addition to these penalties,
FrameAlign also allows you to
specify a separate frameshift penalty
for the creation of gaps that
result in reading frame shifts in the
nucleotide sequence. (See the
ALGORITHM topic for a more detailed
explanation of how gaps are penalized.)
By default, FrameAlign
creates a local
alignment between the
nucleotide and protein
sequences. If you
specify the -GLObal
command-line qualifier,
FrameAlign creates a global alignment
where
gaps are inserted
to optimize the alignment between the
entire
nucleotide sequence and the entire protein sequence.
BLAST
BLAST, or Basic Local Alignment
Search Tool, uses the
method of
Altschul et al. (J. Mol. Biol.
215; 403-410 (1990)) to search
for
similarities between a
query sequence and
all the sequences in a
database. The query sequence and
the database you want to search can
be either protein or nucleic acid in
any combination. The GCG BLAST
program supports five different programs in the BLAST family:
BLASTP, Protein Query Searching a Protein Database
Each database sequence is
compared to the query in a separate
protein-protein pairwise comparison.
BLASTX, Nucleotide Query Searching a Protein Database
The query is
translated, and each
of the six products is
compared to each database sequence in a separate protein-protein
pairwise comparison.
BLASTN, Nucleotide Query Searching a Nucleotide Database
Each database sequence is
compared to the query in a separate
nucleotide-nucleotide pairwise comparison.
TBLASTN, Protein Query Searching a Nucleotide Database
Each nucleotide database sequence is translated, and each of the
six products is
compared to the
query in a
separate
protein-protein pairwise comparison.
TBLASTX, Nucleotide Query Searching a Nucleotide Database
The query and each database sequence are both translated in six
frames, and each of the 12
products is compared in 36 different
pairwise comparisons. This
program is compute-intensive and is
therefore limited to searches of Alu, STS, and EST sequences.
Normally, BLAST decides which BLAST program you want to use simply by
looking at the type (protein or
nucleic acid) of your query sequence
and the database
you have selected. In the case
of
nucleotide-nucleotide searches, there
is more than one program that
can do the search. Adding
-TBLASTX on the command line
means you
want to use the TBLASTX instead of BLASTN.
BLAST either can search databases maintained at your institution (a
local search), or if you are attached to the Internet, it can search
databases maintained by
NCBI (a remote search). Remote
searches
require almost no
resources from your
own computer. More
importantly, the databases at NCBI are updated daily and may be more
current than those maintained locally.
BLAST is a statistically driven
search method that finds regions of
similarity between your query and
a database. Within these
regions
of similarity, called segment
pairs, the sum of the scoring matrix
values of their constituent
symbol pairs is higher than some level
that you would expect to occur by chance alone.
You are prompted to set an
expectation level for the entire
search.
By default this level is 10.0, which means that hits are not reported
unless their scores are above scores that would be expected to occur
more than 10 times by chance alone in the whole search.
FASTA
FastA uses the method of Pearson
and Lipman (Proc. Natl. Acad. Sci.
USA 85; 2444-2448 (1988))
to search for similarities
between one
sequence (the query) and any
group of sequences. In the first step
of this search, the comparison can be viewed as a set of dot plots,
with the query as the vertical sequence and the group of sequences to
which the query
is being compared
as the different horizontal
sequences. This first
step finds the
registers of comparison
(diagonals) having the
largest number of short
perfect matches
(words) for each comparison. In the second
step, these "best"
regions are rescored using a scoring
matrix that allows conservative
replacements, ambiguity symbols, and runs of identities shorter than
the size of a word. In the third
step, the program checks to see if
some of these
initial highest-scoring diagonals
can be joined
together. Finally, the search set sequences with the highest
scores
are aligned to the query sequence for display.
What is a Word?
A word is any short sequence
(n-mer or k-tuple) where you have
set n to some small constant
less than or equal to six.
The
word GGATGG is one of the 4,096
possible words of length 6 that
can be created from an alphabet
consisting of the four letters
G, A, T, and C. The word QL is
one of the 400 possible words of
length 2 that you can make with the 20 letters of the amino acid
alphabet.
TFASTA
TFastA uses the method of Pearson and Lipman (Proc. Natl. Acad. Sci.
USA 85; 2444-2448 (1988)) to
search for similarities between a query
peptide sequence and
any group of nucleotide sequences. TFastA
translates the nucleotide
sequences in all
six frames before
performing the comparison. Each
translated reading frame is treated
as a separate sequence to be searched. In the
first step of this
search, the comparison can be
viewed as a set of dot plots, with the
query as the vertical sequence and the group of sequences to which
the query is being compared as the different horizontal sequences.
This first step finds the
registers of comparison (diagonals) having
the largest number
of short perfect
matches (words) for
each
comparison. In the second
step, these "best" regions are rescored
using a scoring
matrix that allows
conservative replacements,
ambiguity symbols, and runs of
identities shorter than the size of a
word. In the third step, the
program checks to see if some of these
initial highest-scoring diagonals can
be joined together. Finally,
the search set sequences with the
highest scores are aligned to
the
query sequence for display.
What is a Word?
A word is any short sequence
(n-mer or k-tuple) where you have
set n
to some small constant less than or equal to six. The
word GGATGG is one of the 4,096
possible words of length 6 that
can be created from an alphabet consisting of the four letters
G, A, T, and C. The word QL is
one of the 400 possible words of
length 2 that you can make with the 20 letters of the amino acid
alphabet.
FRAMESEARCH
FrameSearch searches a group of
protein sequences for similarity to
one or more nucleotide query
sequences, or searches
a group of
nucleotide sequences for similarity to one or more protein query
sequences. For each sequence comparison, the program
creates the
optimal local alignment of the
best region of similarity between the
protein sequence and all possible codons on
each strand of
the
nucleotide sequence. Because
FrameSearch can match the
protein to
codons in different reading frames of the nucleotide sequence as part
of the same alignment, it can
identify sequence similarity even when
the nucleotide sequence contains reading frame shifts.
In standard sequence alignment
programs, you routinely specify gap
creation and extension penalties.
In addition to these penalties,
FrameSearch also allows you to specify a separate frameshift penalty
for the creation of gaps that result
in reading frame shifts in the
nucleotide sequence. (See the ALGORITHM
topic for a more detailed
explanation of how gaps are penalized.)
By default, the search proceeds as a local alignment
between the
query sequence and each sequence in the search set. Optionally,
you
can search using a global
alignment procedure where
FrameSearch
inserts gaps to optimize the
alignment between the entire nucleotide
sequence and the entire protein sequence.
The search output contains an
ordered list of the sequences in the
search set that have
the highest comparison scores when aligned to
the query sequence. The actual
alignments for these
top-scoring
matches are displayed after the list.
You can specify multiple query
sequences (such as a list file
or a
sequence specification using
an asterisk (*) wildcard) as input to
FrameSearch. The program
compares each query sequence separately
to
the sequences specified in the search set, and it writes a separate
output file for each query
search. If you use a list file as
your
query, you can add begin and
end sequence attributes to
specify the
range for each query
sequence. For more information
about list
files, see "Using List Files
(formerly Files of Sequence
Names)" in
Chapter 2, Using Sequences in the User's Guide.
FINDPATTERNS
FindPatterns locates short sequence patterns. If you are trying to
find a pattern in a sequence or
if you know of a sequence that you
think occurs somewhere within a
larger one, you can find your place
with FindPatterns. FindPatterns
can look through large data sets for
any short sequence patterns you
specify. FindPatterns can recognize
patterns with some symbols mismatched but not with gaps. It supports
the IUB-IUPAC nucleotide
ambiguity codes (see
Appendix III) for
searching through nucleotide sequences.
FindPatterns searches both
strands of a nucleotide
sequence if the
patterns you specify
are not identical on both
strands. If your
sequence is a peptide, FindPatterns searches for
a simple symbol
match between your pattern and the peptide sequence.
FindPatterns names each file on
the screen as it is searched. The
output file shows only sequences where a pattern was found unless you
use the command-line option
-SHOw. Five symbols from the original
sequence are shown on either
side of each "find." The word
/Rev
occurs if the
reverse of the
pattern is found.
If you run
FindPatterns with the
command-line option -NAMes, the output file is
written in list file (formerly called file of sequence names) format,
which you can
use as input to other
Wisconsin Sequence Analysis
Package(TM) programs that support indirect file specifications
When FindPatterns finishes searching for your patterns, it returns to
the first prompt in the program,
FINDPATTERNS in what sequence(s) ?
If you simply press <Return> at the prompt, FindPatterns stops.
FindPatterns keeps writing its results in the same output file (or on
the screen). FindPatterns
prints a short summary on your screen
and
in the output file when the entire session is over.
STRINGSEARCH
Annotations and Definitions
In addition to the actual sequence data, GCG databases contain
two additional types
of data: sequence
annotations, and
definitions.
The annotations contain
the complete documentation for each
entry in the sequence database, including journal and
author
names, sequence features, comments, etc. The annotations appear
at the top of sequences copied from a GCG database with the
Fetch program.
The definitions contain a minimal amount of the annotations
documentation for each entry: the name of the organism, the name
of the gene, the
sequence length, and
usually the date.
Definitions for the GenBank, EMBL, and SWISS-PROT databases also
contain the primary accession number for the sequences. The
definitions are reprinted in the Data Files manual.
The StringSearch program searches through either the definitions
alone or the complete sequence annotations for
text patterns
that you specify. Annotations
take much longer to search than
definitions.
Searching Sequence Definitions
The expression % stringsearch GenBank:* human finds every entry
in the GenBank sequence database
whose definition contains the
text pattern human. The
databases available in addition
to
GenBank are EMBL,
SWISS-PROT, and PIR-Protein. GenEMBL
specifies the sequences in both GenBank and EMBL. Additionally,
definitions searches can be
done on any of
the individual
divisions in GenBank and/or EMBL.
If you believe that a
published human sequence in the
database is 1,531-bases long,
you can search for entries that contain both human and 1531.
When searching definitions, you can specify the set of sequences
you want to search in the same way as for all other Wisconsin
Sequence Analysis Package(TM)
programs with the
following
exception. The specified sequences must
be contained in a
database; you cannot search the definitions of user sequences.
For instance, the
specification Primate:hum* would
search
through the definitions for all
of the sequences in the Primate
division of GenBank that
begin with the pattern hum. You may
also specify the database sequences to search by means of a list
file (formerly called a file of sequence names). Each
sequence
in a list file must be preceded by a logical name for one of the
databases or database divisions.
Sequence specification is
described in detail in Chapter
2, Using Sequences of the User's
Guide.
Searching Complete Sequence Annotations
When you are searching complete sequence annotations, you can
specify the set of sequences you
want to search in the same way
as for all other Wisconsin Package(TM) programs.
Sequence
specification is described
in detail in
Chapter 2, Using
Sequences of the User's Guide.
If your sequence specification is not
preceded by a logical
name, StringSearch looks in all
of the databases and in all of
the GCG data
files to find all possible entry names. The
specification hum* would be
almost the same as GenBank:hum*,
except that if any sequences beginning with hum were present in
databases other than GenBank, they would also be searched.
A
search of all the entries in all the databases takes a very long
time.
Special Considerations for Searching
Keep in mind
that file names
are case sensitive and
database entry
names are case insensitive. Because this
program searches for both file names and database
entry
names, you must
take care when you
enter the character
pattern that makes up your specification.
For example, if you entered Gamma* as a file
specification,
this program would find all entries in the databases whose
names begin with Gamma but no GCG-supplied files would
be
found.
This is because
all the files in the Wisconsin
Package are named using lowercase letters.
Conversely, if
you entered
gamma*, this program would find
all of the
entries
in the databases and all the GCG-supplied files
whose names begin with gamma.
Searching for More Than One Pattern
You can search for more than one text pattern by answering
the
Search for what text
patterns? prompt with a
response like
Human,Globin. StringSearch then
finds all the entries that
contain both human and globin.
You can set StringSearch to show
all the entries that
contain either human or globin
with the
command-line option -MATch=OR.
Specifying Patterns
Blank spaces are removed from the beginning of each pattern
unless that pattern is enclosed in double quotes. For instance,
specifying the pattern Globin shows all entries that contain
globin, while specifying "
Globin" excludes entries
containing
terms like myoglobin in which globin is not preceded by a space.
To specify a double quote
(") as part of a
pattern, use two
double quote marks
(""). To specify a
comma as part of a
pattern, enclose that pattern in quotes.
MAP
Map displays a
sequence that is
being assembled or
analyzed
intensively. Map asks you to enter the names of those enzymes
whose
restriction sites should be marked. If you do not
answer this
question, Map generates
a restriction map
with a representative
isoschizomer from all of the commercially available enzymes. You can
choose to have your sequence translated in any of the six possible
translation frames. You
can also choose to
have only the open
reading frames translated.
After running Map, you
may create a new sequence file with the
peptide sequence from any frame of DNA translation
by using the
ExtractPeptide program with the Map output file.
MAPPLOT
MapPlot is a tool for
genetic engineering. It
helps you visualize
how part of a DNA molecule may
be isolated. MapPlot uses color to
distinguish the types of overhang left after digestion (5' overhangs
are green, 3' overhangs are blue, blunt
ends are black,
and
undetermined overhangs are
red). The site, cut
position, and total
number of cuts are also shown
for each enzyme. The
enzymes that do
not cut are listed below
the plot. You may
choose to plot only
enzymes that have six base
recognition sites or enzymes that cut the
molecule only once.
MAPSORT
MapSort is the best way to predict how the fragments
of an enzyme
digest will look on a gel. You can
tell at a glance which enzymes
cut a molecule in a
given region and whether other
fragments of
similar size could confound isolation of the fragment of interest.
You can concatenate your sequence
with its vector before running
MapSort to see if a single
step isolation is possible and
you can
examine the pattern of
fragments from a multi-enzyme digest. The
sequence can be treated as if it were circular or linear. You can
see which enzymes cut the sequence
only once. Enzymes that cut the
sequence, as well as those that
do not, are shown at the bottom of
the output. The output therefore
contains a complete list of all the
enzymes considered. You
can see the cut sites graphically
with
MapPlot or PlasmidMap.
PLASMIDMAP
PlasmidMap has two purposes:
1) to display and store information
about plasmid constructs;
and 2) to publish or present information
about such constructs.
PlasmidMap reads information about a plasmid from an input file and
produces a labeled circular
plot of that plasmid. The input file
specifies the position of the labels and their styles -- tick, block,
or range. An example of
a suitable input file -- in
this case
containing restriction site information -- is the output file from a
MapSort session run
with the command-line option -PLAsmid.
PlasmidMap can accept more than one labeling file as input, plotting
the information from each file
on the same circular map.
You can
specify more than one file by using a name
containing wildcard
characters or by using a file of filenames (a text file you create
that starts with a line containing two dots (..) and has one filename
per line thereafter). PlasmidMap simply transfers information
from
these labeling files to graph
paper; it does not map the sequence by
itself.
PlasmidMap can draw three kinds of labels:
Ticks
Tick labels are associated with
a single base and are printed
next to a tick around the outside of
the circle. Tick
labels
have been used historically to
identify restriction sites and
coordinates.
Blocks
Block labels are associated with blocks drawn inside the circle.
Blocks can be shaded or left empty.
Ranges
Range labels are associated with lines drawn inside the circle.
Ranges can start and end with arrow heads, arrow tails, blunt
ends, junction heads, and junction tails. The arc for a range
can be narrow or bold.
Every label is defined by a name, a starting coordinate, an ending
coordinate, a strand, a color, and a style (tick, block or range). A
range must also include a
beginning and ending character to indicate
the type of head and tail to draw.
The format of the input file is
described in detail below.
You can set over 30 different
parameters for PlasmidMap.
Therefore,
the values of the parameters are normally defined in an initializing
file rather than by a series of interactive prompts. All parameters
have defaults, so only
parameters that you wish to change need to be
specified in the initializing file.
As with all Wisconsin Sequence Analysis Package(TM) programs, you can
override the values
set in the initializing file
by using
command-line parameters. Values
set on the command line
take
precedence over values in the initializing file.
PRIME
Prime analyzes a template DNA sequence and chooses primer
pairs for
the polymerase chain reaction
(PCR) and primers for DNA sequencing.
For PCR primer pair selection, you can choose a target range of
the
template sequence to be amplified.
For DNA sequencing primers, you
can specify positions on the
template that must be included in the
sequencing.
In selecting appropriate
primers, Prime considers
a variety of
constraints on the primer and
amplified product sequences.
You
either can use
the program's default constraint
values or modify
those values to customize the analysis. You can specify upper and
lower limits for primer and
product melting temperatures and for
primer and product GC contents.
For primers, you can specify a range
of acceptable primer sizes, any
required bases at the 3' end of the
primer (3' clamp),
and a maximum difference in
primer melting
temperatures for PCR primer pairs.
For PCR products, you can specify
a range of acceptable product sizes.
For efficient priming,
you should avoid primers with extensive
self-complementarity in order to
minimize primer secondary structure
and primer dimer formation.
Additionally, in PCR experiments, primer
pairs with extensive complementarity
between the two primers should
be avoided in order to minimize
primer dimer formation. Prime uses
the annealing test
described in the
ALGORITHM topic to
check
individual primers for
self-complementarity and to
check the two
primers in a PCR primer
pair for complementarity to each other.
Using this same annealing test, Prime optionally can screen
against
non-specific primer binding on
the template sequence and on
any
repeated sequences you specify.
The terms forward primer and reverse primer are used in the remainder
of this document and in
the program output. Forward primers are
complementary to sequences on the reverse template strand and create
copies of the
forward strand by primer
extension. Conversely,
reverse primers are
complementary to sequences
on the forward
template strand and
create copies of the reverse strand
by primer
extension.
PILEUP
PileUp creates a multiple sequence alignment using a
simplification
of the progressive alignment method of Feng and Doolittle (Journal of
Molecular Evolution 25; 351-360
(1987)). The method used is similar
to the method described
by Higgins and Sharp (CABIOS
5; 151-153
(1989)).
The multiple alignment procedure
begins with the pairwise alignment
of the two most similar sequences, producing a cluster of two aligned
sequences. This cluster can then
be aligned to the next most related
sequence or cluster of aligned
sequences. Two clusters of sequences
can be aligned by a simple extension of the pairwise alignment of two
individual sequences. The final
alignment is achieved by a series of
progressive, pairwise alignments that include increasingly dissimilar
sequences and clusters, until all sequences have been included in the
final pairwise alignment.
Before alignment, the sequences
are first clustered by similarity to
produce a dendrogram, or tree representation of clustering
relationships. It is this dendrogram
that directs the order of the
subsequent pairwise alignments.
PileUp can plot this dendrogram so
that you can see the order
of the pairwise alignments that created
the final alignment.
As a general rule, PileUp
can align up to 500 sequences,
with any
single sequence in the final alignment
restricted to a maximum length
of 7,000 characters (including gap characters inserted into the
sequence by PileUp to create the alignment). However, if you include
long sequences in the alignment, the number of sequences PileUp can
align decreases. See
the RESTRICTIONS topic, below,
for a
more
complete discussion of sequence number and size limitations.
PRETTY
Pretty prints sequences with
their columns aligned and can display a
consensus for the
alignment, allowing you to look at relationships
among the sequences. This
program can be used for aligned
sequences
in an MSF (multiple sequence
format) file, or for separate sequences
that have had gaps added to make them all align.
You can change the alignments displayed by Pretty with a text editor.
The output from Pretty can then be separated into individual sequence
files by running Pretty with the command line option -UGLy.
PLOTSIMILARITY
PlotSimilarity calculates the average similarity among all members of
a group of aligned sequences at each position in the alignment, using
a user-specified sliding
window of comparison. The window of
comparison is moved along all
sequences, one position at a time, and
the average similarity over the entire window is plotted at
the
middle position of the window.
The average similarity across
the
entire alignment is plotted as a dotted line.
If you give PlotSimilarity a
single input sequence, you can choose
the range and strand for that
sequence, and then
PlotSimilarity
prompts you for the name,
range, and strand of
a second input
sequence. In this way, you can plot the average similarity between
the two aligned sequences created with % gap -OUT.
PROFILEMAKE
There is an
essay on profile analysis in
the Multiple Sequence
Analysis chapter of the Program Manual.
ProfileMake uses the method of
Gribskov, et al (Proc.
Natl. Acad.
Sci. USA 84; 4355-4358 (1987))
to create a profile from a group of
aligned sequences. A profile is a table that contains all of
the
comparison information of a
group of aligned
sequences. These
sequences must be previously
aligned (see the RELATED PROGRAMS topic
below) before running ProfileMake.
The profile contains as many rows
as there are positions in the aligned sequences. Each row contains a
score for the alignment of the corresponding position of the aligned
sequences with each possible base or residue.
The profile is the input
data for ProfileSearch, which
can find
sequences in the database similar to your group of aligned sequences,
and ProfileGap, which
can make an optimal alignment between the
aligned sequences and another sequence.
The aligned sequences
may be specified to
ProfileMake with an
ambiguous file expression or in
a list file similar to the input for
Pretty or LineUp. (See
Chapter 2, Using Sequences in the
User's
Guide for more information.)
PROFILESEARCH
There is an essay
on profile analysis in the Multiple Sequence
Analysis chapter of the Program Manual.
Using the method of Gribskov, et
al. (In Methods in Enzymology, 183;
146-159 (1989)), ProfileSearch accepts a profile from ProfileMake and
uses it to search a database (or
any set of sequences you specify)
for sequences that are similar to the aligned probe sequences used to
create the profile. The algorithm calculates the score (quality)
of
the optimal alignment
between the profile and each sequence in the
database and creates a list of
all of the sequences in the database
with an alignment
score above some threshold.
The results of
ProfileSearch are corrected for compositional effects of the sequence
and for systematic effects of
the sequence length on the score. The
output list can
be displayed as
optimal alignments with
ProfileSegments.
The gap creation
and gap extension
penalties specified for
ProfileSearch are maximum
values. The actual position-specific
gap
penalties at any position are
determined by multiplying the gap
creation penalty by the
percent value in the second to
the last
column of the profile, and the
gap extension penalty by the percent
value in the last column of the profile.
ProfileSearch does a lot
of computing so you will probably want to
run it in the batch queue (see the CONSIDERATIONS topic below).
PROFILESEGMENTS
There is
an essay on
profile analysis in the Multiple Sequence
Analysis chapter of the Program Manual.
ProfileSearch and ProfileSegments
use the method of Gribskov, et
al
(Proc. Natl. Acad.
Sci. USA 84;4355-4358 (1987)). ProfileSearch
compares a profile to a set of sequences and lists the sequences with
similarity to the profile. ProfileSegments then makes an optimal
alignment to display the best segment of similarity in each sequence
in the list. ProfileSegments
uses the alignment procedure of Smith
and Waterman (Advances in
Applied Mathematics 2; 482-489 (1981))
to
search for and align the
segments. The scoring matrix values, gap
creation penalties, and gap extension penalties used to find the best
region of similarity between
the profile and the sequence are all
present in the input file itself and need not be set.
CODONPREFERENCE
CodonPreference finds regions of each reading frame in a DNA sequence
that show either strong
codon preference or unusual
compositional
bias in the third (wobble)
position of each codon. CodonPreference
is useful for locating
protein coding regions,
determining their
reading frames, estimating
the level of expression of a gene, and
locating DNA sequencing errors.
The Preference Curves
The codon preference statistic for each
reading frame shows the
similarity of the codon usage in a
window of that reading frame
to a previously calculated codon usage
table. The statistic
used for the comparison is
described below. The codon
usage
table is a file
of the kind generated by the CodonFrequency
program. A
window of a given size is moved
along the sequence
in increments of one codon (three
bases) and the statistic is
recalculated at every position to make a continuous function.
The statistic for the correct reading
frame of a real gene may
rise significantly above the
background if a suitable codon
frequency table is used and if
the codon usage of the sequence
is strongly biased. Suppress the codon preference curves with
-NOPREFerence.
The Bias Curves
The bias for each reading frame
is the fraction of the
third
position in each codon that is either G or C. The bias of other
nucleotides may be seen by putting an expression on the command
line like -BIAS=AT. Suppress the
bias curves with -NOBIAS.
Errors in Sequence Data
A sequencing error that causes a frame shift may make the curves
for the correct reading frame
fall at the same time that one of
the incorrect frames rises. See
the example below.
The Open Reading Frame Display
Open reading frames
are shown as boxes beneath the plot for
their respective translation frames.
Potential start codons are
shown as short lines that extend
above the top of the box, and
stop codons extend below the bottom of the box. By default,
only the start and stop codons
at the ends of open reading
frames are shown in the frame display.
This can be altered with
the -ALLFrames command-line
option so that all start
and stop
codons are shown. Suppress the
frame display with -NOFRAMes.
The Rare Codon Display
Rare codon choices in each
reading frame are marked below the
open reading frame plot. A codon is considered rare when
its
fraction in the codon frequency
table falls below the threshold
you set with an expression like -RARe=0.1. Suppress the rare
codon display with -NORARe.
Regions of Known Interest
Regions of known interest can be marked
below the x-axis using
the -MARk command line option.
FRAMES
Frames plots open reading frames of a nucleic acid sequences as boxes
bordered by potential start and stop codons. Potential start codons
are shown as short lines that extend above the box and potential stop
codons are shown as
short lines that extend
below the box. By
default, only the start and stop
codons at the ends of open reading
frames are shown in
the frame display; if a stop codon has
been
passed, no stops are shown again until a
start codon is passed; if a
start codon is passed, no
start codons are shown again
until a stop
codon is passed. You can display
all start and stop codons with the
-ALL command-line option. Frames
examines all six translation frames
of a sequence.
If your sequence could have
intervening sequences in it,
use the
command line option -ALL to
get Frames to show all of the possible
start and stop codons in your sequence.
If you know the pattern of
codon usage in the system under
study,
Frames marks the occurrence of rare
codons in each reading frame.
Use the command line option -RARe to have the rare codons marked. If
you want to use a codon
frequency file other than ecohigh.cod, you
can specify this file on the command line with an expression
like
-RARe=codontablename.cod.
REPEAT
Repeat lets you choose a minimum repeat window and stringency and a
search range and then finds all the repeats of at least that size and
stringency within the search range chosen. The repeats are sorted by
position and displayed in an output file as alignments of those parts
of the sequence that make up the
repeats. Repeat tells you
the
number of repeats found for
your settings of window and stringency
before filing the results.
If you feel there are too many repeats,
you may reset
the parameters before writing the
repeats out to a
file. You can limit the number
of repeats shown, or sort the repeats
by quality so that the longest
repeats come at the top of the
list.
See the ALGORITHM topic below to understand precisely what Repeat
does.
COMPOSITION
Composition measures the composition of one or a group of sequences.
If you specify only one
sequence, you can choose a range within
the
sequence. Lowercase letters are converted to uppercase and counted
with their uppercase
equivalents. If you
specify a group
of
sequences, Composition displays
the name of each
sequence as it
finishes the measurement for that sequence.
TERMINATOR
Terminator uses a table of
the dinucleotide frequencies for each
position from a set of known
terminators to find places
in a new
sequence where terminator-like sequences occur. Terminator finds all
discrete examples in the
searched sequence where a measurement falls
above some user-defined
threshold value. The measurement for each
alignment of the table over the
sequence is the sum of the values in
the table for each dinucleotide from the sequence. The method can
also restrict the set of terminatorlike sequences shown to those that
fall above some threshold for the presence of a GC-rich dyad symmetry
near the poly-U region.
The method used by Terminator is
described in detail in two papers:
Brendel, V. and Trifonov, E. N., Nucl. Acids Res. 12 4411-4427 (1984)
and Brendel, V. and Trifonov, E. N. in CODATA Conference Proceedings,
Jerusalem, 1984. Any use of
Terminator that results in publication
should cite these papers.
CODONFREQUENCY
CodonFrequency counts codons and writes their frequencies into codon
frequency tables. It counts the codons from ranges within sequences
or existing codon frequency
tables. The output table is a file with
the sum of all the observations
for each of the 64 possible codons.
This file is suitable for input to other GCG programs, including
BackTranslate, CodonPreference, and Correspond.
CodonFrequency supports the
assembly of fragments
from circular
molecules by letting you define a range in the sequence that extends
across the end and into the
beginning of a molecule. The terminal
bell rings when a circular range is chosen.
To count codons from
sequences, specify ranges
until you have
assembled a sequence
you want to
count. For each
range,
CodonFrequency shows you the
starting and ending symbols to double
check that you have chosen the range and strand accurately.
After choosing each range, you must decide if you would like to
add
another exon to the gene
or count the codons in the gene
you have
assembled. It is critical that
you count the codon frequencies from
multi-exon genes after
assembling all the ranges since intervening
sequences often interrupt such genes within a codon, thus destroying
the reading frame.
After CodonFrequency counts all
the codons in your
gene, you may
specify another gene from
the current sequence file or
get other
sequence files or codon frequency tables.
You can specify multiple sequences (such as a list file or sequence
specification using an asterisk
(*) wildcard) to count codons from
more than one sequence at a time.
By default, each sequence in a
multiple sequence
specification is treated as a separate
gene and
counted separately by CodonFrequency. If you add the -ONEPEPtide
command-line qualifier, then
all sequences in a multiple
sequence
specification are concatentated together into a single
sequence
before counting codons. If you use a list file to specify multiple
sequences, you can add begin,
end, and strand sequence attributes to
specify the range and strand for each sequence. For more information
about list files, see "Using List
Files (formerly Files of
Sequence
Names)" in Chapter 2, Using Sequences in the User's Guide.
After each sequence range is
counted or each new
codon frequency
table is read, CodonFrequency asks if you want to write the data to a
file. If you choose to do so, the program writes a
file with the
number of observations for each codon.
In addition, CodonFrequency
normalizes the codon observations
to a frequency per thousand and to
a fraction for each codon within its synonymous family.
MOTIFS
Motifs looks for protein sequence
motifs by checking your protein
sequence for every
sequence pattern in
the PROSITE Dictionary.
Motifs can recognize
the patterns with
some of the
symbols
mismatched, but not with gaps.
Currently, Motifs can only search for
patterns in protein sequences.
There is a very informative
abstract on every motif in the PROSITE
Dictionary. These abstracts are
displayed next to any motif found in
your sequence.
The PROSITE Dictionary
was written by Dr. Amos
Bairoch of the
University of Geneva -- a labor of love.
PEPTIDESORT
PeptideSort cuts a
peptide sequence with
any or all of the
proteolytic enzymes and reagents
listed in the public or local data
file proenzall.dat. The peptides
from each digest are sorted
by
position, weight, and
retention time in a
high-pressure liquid
chromatograph at pH 2.1. For
each peptide in
each sorting, the
following data are
displayed: beginning and
ending positions,
molecular weight, HPLC retention at pH 2.1, HPLC retention at pH 7.4,
charge, number of aromatic residues, number of acidic
residues,
number of basic
residues, number of residues containing
sulfur,
number of hydrophilic residues, and number of hydrophobic residues.
The content, isoelectric point,
and molar extinction coefficient
at
280 nm of each peptide are shown with the table of peptides sorted by
position. The content can
be displayed in the order
of expected
elution from an amino acid analyzer.
PEPTIDEMAP
PeptideMap marks a peptide sequence at every position where a known
proteolytic enzyme or reagent might cut it. You can select one or a
few enzymes or let PeptideMap use the whole list.
PeptideMap is simply the program Map run with the command-line option
-PROGRAMname=PeptideMap.
(See the documentation for Map in the
Program Manual for a complete description.)
HELICALWHEEL
HelicalWheel plots a
helical wheel representation of a peptide
sequence. Each residue is
offset from the preceding one by
100
degrees, the typical angle of rotation for an alpha-helix. The angle
of rotation can be changed using the
-ANGle and -BETa command-line
options.
MFOLD
MFold is an adaptation of the
mfold package by Zuker and Jaeger that
has been modified to
work with the Wisconsin
Package(TM). Their
method uses the energy
rules developed by Turner and colleagues to
determine optimal and suboptimal secondary structures
for an RNA
molecule. (See the ACKNOWLEDGEMENTS
topic for references.)
Using energy minimization criteria, any predicted "optimal"
secondary
structure for an RNA molecule depends on the model of RNA folding and
the specific folding energies
used to calculate that
structure.
Different optimal foldings may be calculated if the folding energies
are changed even slightly. Because of uncertainties in the folding
model and the folding energies,
the "correct" folding may not be the
"optimal" folding determined by the program. You may
therefore want
to view many optimal and
suboptimal structures within a
few percent
of the minimum energy. You can
use the variation
among these
structures to determine which
regions of the secondary structure you
can predict reliably. For instance, a region of
the RNA molecule
containing the same helix in most calculated optimal and
suboptimal
secondary structures may be
more reliably predicted
than other
regions with greater variation.
MFold calculates energy matrices that determine all optimal
and
suboptimal secondary structures for
an RNA molecule. The
program
writes these energy matrices to an output file. A companion program,
PlotFold, reads this output file and displays a representative set of
optimal and suboptimal
secondary structures for the RNA
molecule,
within any increment of the computed minimum free energy you choose.
You can choose any of
several different graphic
representations for
displaying the secondary structures in PlotFold.
PLOTFOLD
MFold uses the method of Zuker
(see the MFold entry in this manual
for more information) to determine optimal and suboptimal secondary
structures for an
RNA molecule. MFold
writes an output
file
containing energy matrices that
determine all optimal and suboptimal
foldings for the RNA molecule.
PlotFold reads this output file and
displays representations of the optimal and suboptimal foldings.
PlotFold allows you
to choose from
among seven different
representations of the optimal and
suboptimal foldings determined by
MFold. Each of
these options allows you to
specify an energy
increment, which is the
highest deviation in
kcal/mole from the
computed free energy minimum that a structure can have in order to be
plotted. For example,
if the predicted optimal
folding has a
computed free energy
of -114.5 kcal/mole, and
you select a
5.7 kcal/mole energy increment, then all plotted structures must have
calculated free energies
no greater than
-108.8 kcal/mole
(-114.5 + 5.7 = -108.8).
The first two folding
representations, the energy dotplot and the
p-num plot, plot
base pairing information
from all secondary
structures that have free energies within the energy increment you
specify. You can use these
displays to determine which regions of
the secondary structure
prediction are well defined (see the example
sessions below).
Energy Dotplot (menu option A)
The energy dotplot
indicates all of the base pairs
involved in
all optimal and suboptimal
secondary structures within
the
energy increment you
specify. The plot takes the form of a
two-dimensional graph where both axes of the graph represent the
same RNA sequence. Each
point drawn in the graph
indicates a
base pair between the ribonucleotides whose
positions in the
sequence are the coordinates of that point on the graph.
P-Num Plot (menu option B)
The p-num plot graphs the amount
of variability in pairing at
each position in the RNA molecule. For each position of the
sequence along the horizontal
axis, the height of the
plot
indicates how many different pairing partners the program finds
in all predicted foldings
within the energy
increment you
specify.
The remaining six PlotFold
options plot a
sampling of specific
secondary structures that have
calculated free energies within the
energy increment you
specify. These remaining options all display
the same information, but in different forms. Rather than attempting
to plot each
secondary structure whose computed free energy falls
within the energy increment, PlotFold selects representative foldings
that are sufficiently different
from each other and are still within
the specified energy
increment. You can specify how different each
folding must be from the others in response to the
window size
program prompt. To understand the concept of a window size,
we must
first define the idea of a
distance between base pairs in
different
foldings. The distance
between two base
pairs, r(i)-r(j) and
r(i')-r(j') is the greater
of |i - i'| and |j - j'|. Each
listed
folding must have at least a window size number of base pairs that
are greater than window
size distance from the base
pairs in any
other listed folding.
Each of these five PlotFold
options also lets you select the number
of structures to plot. Since the
number of structures that meet both
the energy increment and window size criteria may be less than your
selection, fewer secondary structures may actually be plotted.
Circles Plot (menu option C)
The circles plot makes a
circular Nussinov plot
of an RNA
secondary structure. The
circular graph represents the sequence
as a segment of a
circle. You can set the radius and
the
angular width of one base
so that plots of different secondary
structures are strictly comparable. Arcs
or chords connect
paired bases; hairpin, bulge, interior,
and multibranched loops
are easily seen.
Domes Plot (menu option D)
The domes plot represents a
folded RNA sequence as a line
with
elliptical arcs connecting paired bases. This representation
has the property that the length
of the arc is proportional to
the distance (along the primary structure) between
the bases;
all loops are easily seen.
Mountains Plot (menu option E)
The mountains plot makes a graph that looks
like a mountain
range. Horizontal striations
upon a particular peak are bonds
between paired bases, and vertical
links between the horizontal
striations represent stems.
Squiggles Plot (menu option F)
The squiggles plot is a representation similar to what you might
draw by hand; that is, bonds formed between bases are drawn as
chords. Bases are shown
participating in stems, as well as in
hairpin, bulge, interior, and multipbranched loops.
Text Output (menu option G)
The text output representation of the RNA secondary structure is
similar to the squiggles plot,
but you don't need a
graphics
device to see it. The output is
written into a text file that
you can view with any text editor.
If you exclude a region of
the RNA molecule
from folding in
MFold with either
the
-CLOSedexcise or -OPENexcise option, you can view the predicted
secondary structures only as text output; do not use any of the
graphic plotting options of PlotFold to display the results.
Connect File Output (menu option H)
The Connect file is a
base-by-base text output file of
optimal
and suboptimal RNA secondary structures. This file can be used
as input to
several programs that
produce graphical
representations of RNA secondary
structures. In the Wisconsin
Package(TM), the Squiggles,
Circles, Domes, and
Mountains
programs can read the Connect file
output of MFold and display
the secondary structure of the
optimal structure, only. Other
publicly available RNA secondary
structure display programs may
be able to
display all optimal
and suboptimal secondary
structures listed in the Connect file.
The examples below
demonstrate three different PlotFold
options for
the secondary structure representations.
FOLDRNA
FoldRNA finds a secondary structure of minimum free energy for an RNA
molecule based on published values of stacking and loop destabilizing
energies. FoldRNA is the
program of Michael
Zuker (Methods in
Enzymology, 180, 262-288(1989).
The energies used by Zuker's program
were first described by Winston
Salser (CSHSQB 42; 985) and are now
defined by Turner
(Freier et al., Proc. Natl. Acad. Sci. USA 83:
9373-9377 (1986)).
You should be
aware of the
limitations of energy
minimizing
algorithms in predicting
real secondary structures. The
structure
reported in the output file is only
one of a family of
structures
that have the
same or nearly
the same energy. The
number of
structures that have
similar energies to
the optimal structure
reported by FoldRNA may be very
large when several hundred bases are
folded or when the secondary structure is not strong.
SQUIGGLES
The program FoldRNA calculates an optimal RNA secondary structure and
writes a base-by-base output file with the sequence name and the file
name extension .connect. The
Circles, Domes, DotPlot, Mountains, and
Squiggles programs accept the
FoldRNA output file and graph the RNA
secondary structure. Circles, Domes, DotPlot, and Mountains make
abstract representations, but
the graphs they draw are
easier to
compare if you are looking for secondary structure motifs.
Squiggles creates a representation similar to what you might draw by
hand; that is, bonds formed between
bases are drawn as chords. The
entire structure looks like
an airport or intersecting railroad
tracks. The spider-like
graph plotted by Squiggles represents
the
sequence as bases connected by chords.
Bases are shown participating
in stems, as well
as in hairpin, bulge, interior and bifurcation
loops. These structures are
easily seen in Squiggles.
To make your graph
more readable, you can label the bases, show
sequence numbers at set
intervals, and pivot up to nine
stems. You
can also change the character height for the number and base labels.
STEMLOOP
StemLoop searches for inverted repeats in your sequence after
you
choose a minimum stem length and minimum and maximum loop sizes. You
must also specify a minimum number of bonds per stem with G-T,
A-T,
and G-C scored as 1, 2, and
3 bonds, respectively. The stems found
can be sorted by position, size
(stem length), or quality (number of
bonds) and can be either filed or displayed on the screen. StemLoop
tells you the number of stems found for your settings of minimum stem
size, maximum loop size, minimum loop size, and minimum bonds per
stem. If you
feel there are too many
stems, you may reset the
parameters without reviewing the
stems found or view only the best
stems found. To view only the
best stems, there must be more than 25
stems found and you must
sort them by quality or size. (See the
ALGORITHM topic below to understand precisely what StemLoop does.)
REFORMAT
Reformat rewrites sequence files to make them usable by the Wisconsin
Sequence Analysis
Package(TM) or to alter their appearance. The
following are some of the manipulations that Reformat can perform:
- converting sequence files that
were prepared or edited with a text
editor or transferred to your computer from
another computer
into GCG format.
- converting between
multiple sequence file
(MSF) format and
individual sequences in GCG format.
- correcting the sequence type
(protein or nucleic acid) of sequence
files that have no type or that were incorrectly typed when they
were created.
- converting nucleic acid
sequences between DNA (T, t) and RNA (U,
u) representations.
- converting peptide
sequences between one-letter and three-letter
amino acid representations.
- converting sequences to all
uppercase or all lowercase characters.
- removing gap characters from
sequence files.
Reformat can also be used to
rewrite into GCG format MSF files that
you've edited with a text editor.
In order to use Reformat on
sequence files, the files must contain a
heading, a dividing line,
and a sequence, as described below. You
can use a text editor to make
your "foreign" sequence files
conform
to this arrangement.
FROMEMBL
Use FromEMBL when you want to
use sequences in EMBL's distribution
format with the Wisconsin Sequence Analysis Package(TM). Since EMBL
maintains many sequences in one file, FromEMBL must write many output
files, one for each sequence in
the EMBL file. Each output file is
named according to the identifier word on the ID
line at the
beginning of each sequence entry.
All documentation from the EMBL
input files is preserved in the GCG
output files. The nucleic acid
ambiguity codes are preserved except that the hyphen (-)
symbol in
the EMBL sequences is changed to N in the GCG files.
FROMGENBANK
Use FromGenBank when you want to
use sequences in the GenBank flat
file distribution format
with the Wisconsin
Sequence Analysis
Package(TM). Since GenBank maintains many sequences in one
file,
FromGenBank must write many
output files, one for each sequence in
the GenBank file. Each
output file is
named according to the
identifier word on the
LOCUS line at the beginning of
each sequence
entry. All the
documentation from the
GenBank input files is
preserved in the GCG output files.
FROMFASTA
Use FromFastA when you want to
convert sequences that are in FastA
format into a format suitable
for use with programs in the Wisconsin
Sequence Analysis Package(TM). FastA format may
maintain many
sequences in one file; in such a case FromFastA writes many output
files, one for each sequence in the FastA file. Each output file is
named according to the first
word (following the > character) on the
documention line just above the sequence data in the FastA file. The
documentation line from the FastA
input file(s) is preserved in
the
GCG output file(s).
FromFastA can convert sets of
FastA sequence
files that are specified with a file of filenames or with multiple
file specification syntax.
TOSTADEN
Any sequence file in GCG format can be converted with ToStaden into a
format suitable for use in the Staden programs. If the sequence is a
nucleic acid sequence, the compatible
ambiguity codes are converted
from
the IUB-IUPAC versions to
Staden's versions. You can see how
they are converted in Appendix III or in the example below.
TOFASTA
Sequences in GCG format can be
converted into a format suitable for
use by programs that require sequences in FastA format. ToFastA
accepts one or more GCG sequences as input and by default creates one
output file containing all the sequences in FastA format. However,
NCBI's BLAST family of programs accepts only one sequence per input
file. Therefore, if you put
-BLAst on the command
line, ToFastA
writes your output into separate files,
naming each output file with
the input sequence's name and the filename extension .tfa.
TRANSLATE
Translate creates a peptide sequence
by translating nucleic acid
sequences that you specify. In addition to translating a single
range of a given nucleotide sequence, it can concatenate exons into a
single assembly for translation.
The exons can be of any length, can
come from either strand,
and can come
from more than one sequence
file. Unlike most Wisconsin
Package(TM) programs, Translate lets you
specify ranges that extend across the end and into the beginning of a
sequence. The terminal bell
rings when a circular range is chosen.
Translate can be run either interactively or noninteractively. When
you specify a single sequence to translate and -Default is not on the
command line, it works interactively,
prompting you for each segment
to translate. To run Translate
noninteractively, either use
the
-Default command-line switch or supply a multiple file specification
for the input file by
means of a wild card file specification or a
list file. (See the
INPUT FILE topic
below for more detailed
information.)
Translate supports the IUB-IUPAC character set for the representation
of nucleotide ambiguity. See Appendix III for a list of the
IUB
codes and their meanings.
BACKTRANSLATE
BackTranslate uses a peptide sequence and a codon usage file to make
a table of possible
backtranslations. The program
can write all of
the codons for each amino acid in the peptide sequence,
showing the
frequency of each within its synonymous group.
BackTranslate also calculates either the most
probable
back-translation or the fully
ambiguous depiction of the nucleotide
sequence. In either case, it writes the implied sequence such that
the output file can be used as the input file for other sequence
mapping or comparison programs.
In the table of back-translations
options, the codons are written in
order of their preference
in the codon frequency
table. Below each
block of synonymous codons, there is a number between 0 and 1,000; it
is the product of the probabilities for the most likely codon for the
next four amino acids multiplied by 1,000. The higher the
number,
the more likely it
is that the next 12
nucleotides contain the
most-preferred codons. All
of the codons and their preferences are
included to help
you look critically at the alternative
oligonucleotides that you might want to synthesize. For instance, if
for one amino acid the codon preference is not strong,
you could
consider making a
mixture that contains
all of the different
possibilities.
PUBLISH
Publish creates a text file that
you can customize for publication.
You can choose lines that
represent the sequence, its complement,
a
decimal scale, a three-letter translation, a one-letter translation,
a numbering line with numbers
at every twentieth symbol,
a blank
line, or a tagged line. Additional
sequences can be shown either
completely or with only the differences marked. A match line between
any two sequence lines
marks the matches. The lines can appear in
any order. Publish can number
each line starting from any number you
choose. The line is numbered at
both ends if you select the line
from the menu by typing its menu
letter in uppercase. Each type of
line is described in detail below.
The output can be blocked using
two parameters: the block size, and the number of blocks per line.
The ranges of translation must be chosen by you for each translation
line selected. You can
translate as many non-overlapping
ranges as
you want for
each translation line
you select. If
you have
overlapping ranges of
translation, you must
select two translation
lines and set the ranges appropriately.
Acknowledgement
A few pieces of this
document are from the very good manual written by Hans Mehlin, Karolinska
Institute, Stockholm and which is available at http://kisac.cmb.ki.se/senn/files.html.