Άσκηση Εξέλιξης 1
Πολλαπλή ευθυγράμμιση Ακολουθιώv και κατασκευή
Φυλογενετικών δέντρων.
(CLUSTAL: Multiple Alignment and
Phylogenetic Trees)
Πρόγραμμα από:
Des Higgins
European Molecular Biology Laboratory
Postfach 10.2209
D-6900 Heidelberg
Germany.
Δημοσιευμένες αναφορές:
Higgins, D.G. and Sharp, Ρ.Μ. (1988) CLUSTAL: α package for
performing multiple sequence alignments οn α microcomputer. Gene
73, 237-244.
Higgins, D.G. and Sharp, Ρ.Μ. (1989) Fast and sensitive multiple
sequence alignments οn α microcomputer.
CABIOS 5, 151-153.
Copyright (C)
D.Higgins, 1991
Αll rights reserved
ΕΙΣΑΓΩΓΗ.
Στην παρακολούθηση της εξέλιξης ακολουθιών βάσεων και αμινοξέων
είναι απαραίτητη η ευθυγράμμιση (alignment) των ακολουθιών με τον καλλίτερο
δυνατό τρόπο (ομοιότητες,συγγένειες αμινοξέων κλπ.). Είναι επίσης απαραίτητο
στάδιο στην εύρεση της γενετικής απόστασης μεταξύ οργανισμών, την αναγνώριση
ειδικών περιοχών που σχετίζονται με την δράση των μορίων και την κατασκευή των
φυλογενετικών δέντρων.
Το πρόγραμμα CLUSTAL αυτόματα ευθυγραμμίζει ακολουθίες βάσεων ή
αμινοξέων, όταν αυτές έχουν κάποια συγγένεια (>10%), με την εισαγωγή κενών
θέσεων ώπου είναι απαραίτητες και προτείνει ποσοστιαίες αποστάσεις μεταξύ των
ακολουθιών βασισμένες στον αριθμό και το είδος των μεταλλαγών που
παρατηρούνται.
Τα δεδομένα είναι της μορφής NBRF/PIR (βάσεων δεδομένων
αμινοξικών/νουκλεινικών ακολουθιών) με ένα γράμμα για κάθε αμινοξύ/νουκλ. βάση.
Στην έξοδο το πρόγραμμα CLUSTAL μας δίνει τις αμινοξικές
ακολουθίες ευθυγραμμισμένες τονίζοντας την τυχόν απόλυτη διατήρηση αμινοξέων
και αν ζητηθεί πίνακα σύνδεσης για την κατασκευή φυλογενετικού δέντρου.
Για την επιλογή 3 εμφανίζεται ο παρακάτω πίνακας επιλογής:
******Profile*Alignment*Menu******
1. Input lst. profile/sequence
2. Input 2nd. profile/sequence
3. Do alignment nοw
4. Alignment parameters
5. Output format options
S. Execute a system command
Η. HELP
or press [RETURN] to go back to main menu
Your choice:
Αυτές οι επιλογές χρησιμοποιούνται μόνον εάν έχουμε προηγουμένως
κάνει ευθυγράμμιση για κάποια υποομάδα ακολουθιών.
Για την επιλογή 4 του φυλογενετικού δέντρου εμφανίζεται ο παρακάτω
πίνακας επιλογής:
******Phylogenetic*tree*Menu******
1. Input an alignment
2. Exclude positions with gaps? = OFF
3. Correct for multiple substitutions? =
OFF
4. Draw tree now
5. Bootstrap tree
S. Execute a system command
Η. HELP
or press [RETURNJ to go back to main menu
Your choice:
Με την επιλογή 1 διαβάζουμε ένα αρχείο με προηγούμενη
ευθυγράμμιση.
Με την επιλογή 2 εξαιρούμε περιοχές που έχουν γίνει διαγραφές ή
πρόσθετη εισαγωγή αμινοξέων ή βάσεων (deletion and insertion).
Με την επιλογή 3 μπορούμε να διορθώσουμε την απόκλιση για
πολλαπλές μεταλλαγές.
Με την επιλογή 4 υπολογίζεται το φυλογενετικο δέντρο. Αυτό ανάλογα
με τον αριθμό των ακολουθιών είναι αρκετά χρονοβόρο.
Με το [Enter το πρόγραμμα οδηγείται στο προηγούμενο πίνακα
επιλογής ενώ από τον αρχικό πίνακα επιλογής.το Χ οδηγεί στο τέλος του
προγράμματος και επιστροφή στο λειτουργικό σύστημα DOS.
ΠΕΡΙΓΡΑΦΗ ΤΗΣ ΑΣΚΗΣΗΣ
Δίνονται 23 αμινοξικές ακολουθίες του κυτοχρώματος c στο αρχείο
ALL.SEQ .
α. Χρησιμοποιώντας την μέθοδο πολλαπλής ευθυγράμμισης κατασκευάστε
πρόχειρο δενδρόγραμμα με την μέθοδο UPGMA, ευθυγραμμίστε τις 23 ακολουθίες,
τυπώστε τις και αναγνωρίσατε τις περιοχές μετάλλαξης.
β.Υπολογίστε το φυλογεννετικό δέντρο και σχεδιάστε το με και χωρίς
διόρθωση για πολλαπλές μεταλλάξεις. Παρατηρείτε διαφορές;
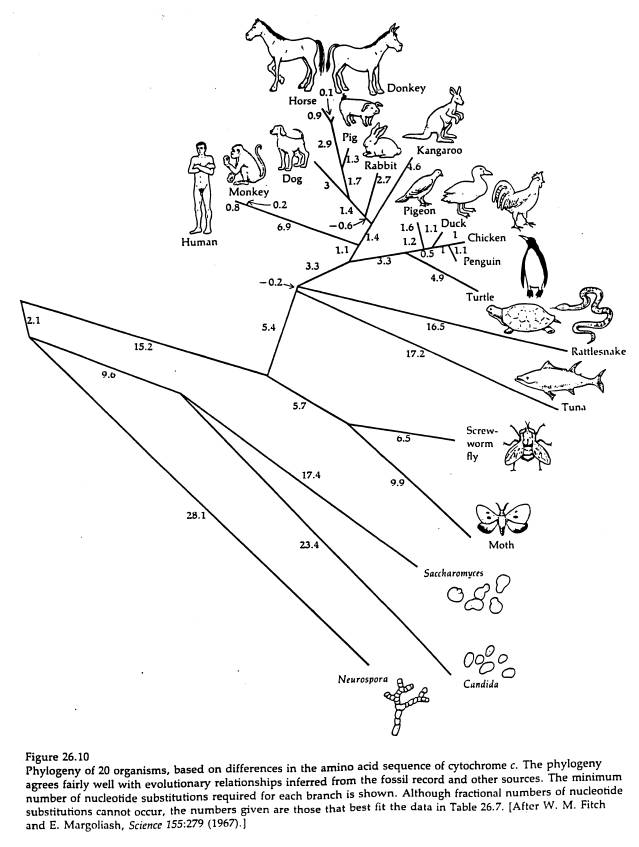
γ. Συγκρίνετε το φυλογενετικό δέντρο των ακολουθιών με αυτό που
δίνεται για την εξέλιξη 20 ειδών. Προτείνετε αντιστοιχία κωδικού πρωτείνης και
είδους.
δ. Πόσες μεταλλαγές βάσεων έχουμε μεταξύ κυτοχρωμάτων C ανθρώπου
και αλόγου; (Δίνεται πίνακας αντιστοίχησης αμινοξέων και νουκλεινικών βάσεων.)
ΒΙΒΛΙΟΓΡΑΦΙΑ
Dayhoff, Μ.Ο., Schwartz, R.M.
and Orcutt, B.C. (1978) in Atlas of Protein Sequence and Structure, Vol. 5
supplement 3, Dayhoff, Μ.Ο. (ed.), NBRF, Washington, p. 345.
Felsenstein, J. (1985) Confidence limits οn phylogenies: an approach using the
bootstrap. Evolution 39, 783-791.
Feng, D.-F. and Doolittle, R.F. (1987)
Progressive sequence alignment as a prerequisite to correct phylogenetic trees.
J.Mol.Evol. 25, 351-360.
Gotoh, O. (1982) An improved algorithm for
matching biological sequences. J.Mol.Biol. 162, 705-708.
Gribskov, Μ., McLachlan, A.D. and Eisenberg, D. (1987) Profile analysis: detection
of distantly related proteins. PNAS USA 84 , 4355-4358.
Higgins, D.G. and Sharp, Ρ.Μ. (1988) CLUSTAL: a package for performing multiple sequence alignments
on a microcomputer. Gene 73, 237-244.
Higgins, D.G. and Sharp, Ρ.Μ. (1989) Fast and sensitive multiple sequence alignments οπ a microcomputer. CABIOS 5, 151-153.
Kimura, Μ. (1980) Α simple method for
estimating evolutionary rates of base substitutions through comparative studies
of nucleotide sequences. J. Μοl. Ενοl. 16, 111-120.
Kimura, Μ. (1983) The Neutral Theory of Molecular Evolution. Cambridge University
Press, Cambridge, England.
Li, W.-Η., Wu, C.-Ι. and Luo, C.-C.
(1985) Α new method for
estimating synonymous and nonsynonymous rates of nucleotide substitution
considering the relative likelihood of nucleotide and codon changes.
Mol.Biol.Evol. 2, 150-174.
Myers, E.W. and Miller, W. (1988) Optimal
alignments in linear space. CABIOS 4, 11-17.
Pearson, W.R. and Lipman, D.J. (1988)
Improved tools for biological sequence comparison. PNAS USA 85, 2444-2448.
Saitou, Ν. and Nei, Μ. (1987) The
neighbor-joining method: a new method for reconstructing phylogenetic trees.
Mol.Biol.Evol. 4, 406-425.
Sneath,'Ρ.Η.Α. and Sokal, R.R. (1973) Numerical
Taxonomy. Freeman, San Francisco.
Sokal, R.R. and Michener, C.D. (1958) Α statistical method forevaluating
systematic relationships. Univ.Kansas Sci.Bull. 38,1409-1438.
Vingron, Μ. and Argos, Ρ. (1991) Motif
recognition and alignmentfor many sequences by comparison of dot matrices.
J.Mol.Biol. 218,33-43.
Wilbur, W.J. and Lipman, D.J. (1983) Rapid
similarity searches of nucleic acid and protein data banks. PNAS USA 80,
726-730.
Πίνακας συμβολισμού των 20 αμινοξέων και
των κωδικονίων που τα μεταγράφουν.
|
ΑΜΙΝΟΞΕΑ |
ΣΥΜΒΟΛΑ
|
ΚΩΔΙΚΟΝΙΑ
|
||||||
|
Ασπαραγινικό οξύ |
Asp |
D |
GAC |
GAU |
|
|
|
|
|
Γλουταμινικό οξύ |
Glu |
E |
GAA |
GAG |
|
|
|
|
|
Αργινίνη |
Arg |
R |
AGA |
AGG |
CGA |
CGC |
CGG |
CGU |
|
Λυσίνη |
Lys |
K |
AAA |
AAG |
|
|
|
|
|
Ιστιδίνη |
His |
H |
CAC |
CAU |
|
|
|
|
|
Ασπαραγίνη |
Asn |
N |
AAC |
AAU |
|
|
|
|
|
Γλουταμίνη |
Gln |
Q |
CAA |
CAG |
|
|
|
|
|
Σερίνη |
Ser |
S |
AGC |
AGU |
UCA |
UCC |
UCG |
UCU |
|
Θρεονίνη |
Thr |
T |
ACA |
ACC |
ACG |
ACU |
|
|
|
Τυροσίνη |
Tyr |
Y |
UAC |
UAU |
|
|
|
|
|
Αλανίνη |
Ala |
A |
GCA |
GCC |
GCG |
GCU |
|
|
|
Γλυκίνη |
Gly |
G |
GGA |
GGC |
GGG |
GGU |
|
|
|
Βαλίνη |
Val |
V |
GUA |
GUC |
GUG |
GUU |
|
|
|
Λευκίνη |
Leu |
L |
UUA |
UUG |
CUA |
CUC |
CUG |
CUU |
|
Ισολευκίνη |
Ile |
I |
AUA |
AUC |
AUU |
|
|
|
|
Προλίνη |
Pro |
P |
CCA |
CCC |
CCG |
CCU |
|
|
|
Φαινυλαλανίνη |
Phe |
F |
UUC |
UUU |
|
|
|
|
|
Μεθειονίνη |
Met |
M |
AUG |
|
|
|
|
|
|
Τρυπτοφάνη |
Trp |
W |
UGG |
|
|
|
|
|
|
Κυστείνη |
Cys |
C |
UGC |
UGU |
|
|
|
|
|
Κωδικόνια Τερματισμού |
UAA UAG UGA |
Πίνακας αναπαράστασης φυλογένειας ειδών
που είναι γνωστή η αμινοξική ακολουθία του κυτοχρώματος C
Πολλαπλή ευθυγράμμιση ακολουθιών με το
πρόγραμμα CLUSTAL
CLUSTAL V multiple sequence alignment
CCHU
-----------GDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRK
CCMQR
-----------GDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRK
CCHO
-----------GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLHGLFGRK
CCHOD
-----------GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLHGLFGRK
CCPG
-----------GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLHGLFGRK
CCRB
-----------GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLHGLFGRK
CCDG
-----------GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLHGLFGRK
CCKGG
-----------GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLNGIFGRK
CCPY
-----------GDIEKGKKIFVQKCSQCHTVEKGGKHKTGPNLHGLFGRK
CCCH
-----------GDIEKGKKIFVQKCSQCHTVEKGGKHKTGPNLHGLFGRK
CCDK
-----------GDVEKGKKIFVQKCSQCHTVEKGGKHKTGPNLHGLFGRK
CCPN
-----------GDIEKGKKIFVQKCSQCHTVEKGGKHKTGPNLHGIFGRK
CCST
-----------GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLNGLIGRK
CCRS
-----------GDVEKGKKIFSMKCGTCHTVEEGGKHKTGPNLHGLFGRK
CCBN
-----------GDVAKGKKTFVQKCAQCHTVENGGKHKVGPNLWGLFGRK
CCFFCM
-------GVPAGDVEKGKKLFVQRCAQCHTVEAGGKHKVGPNLHGLIGRK
CCNC
-------GFSAGDSKKGANLFKTRCAQCHTLEEGGGNKIGPALHGLFGRK
CCRZ --ASFS-EAPPGNPKAGEKIFKTKCAQCHTVDKGAGHKQGPNLNGLFGRQ
CCPO
--ASFG-EAPPGNPKAGEKIFKTKCAQCHTVDKGAGHKEGPNLNGLFGRQ
CCRPBO
--ASFD-EAPPGNSKAGEKIFKTKCAQCHTVDKGAGHKQGPNLNGLFGRQ
CCWT
--ASFS-EAPPGNPDAGAKIFKTKCAQCHTVDAGAGHKQGPNLHGLFGRQ
CCCRCO
PPKARE-PLPPGDAAKGEKIFKGRAAQCHTGAKGGANGVGPNLFGIVNRH
CCBY
-----T-EFKAGSAKKGATLFKTRCLQCHTVEKGGPHKVGPNLHGIFGRH
CCTE
GPKEPEVTVPEGDASAGRDIFDSQCSACHAIE--GDSTAAPVLGGVIGRK
*
* * . **. .
.* * *. *
CCHU
TGQAPGYSYTAANKNKGIIWGEDTLMEYLENPKKYIPGTKMIFVGIKKKE
CCMQR
TGQAPGYSYTAANKNKGITWGEDTLMEYLENPKKYIPGTKMIFVGIKKKE
CCHO
TGQAPGFTYTDANKNKGITWKEETLMEYLENPKKYIPGTKMIFAGIKKKT
CCHOD
TGQAPGFSYTDANKNKGITWKEETLMEYLENPKKYIPGTKMIFAGIKKKT
CCPG
TGQAPGFSYTDANKNKGITWGEETLMEYLENPKKYIPGTKMIFAGIKKKG
CCRB
TGQAVGFSYTDANKNKGITWGEDTLMEYLENPKKYIPGTKMIFAGIKKKD
CCDG
TGQAPGFSYTDANKNKGITWGEETLMEYLENPKKYIPGTKMIFAGIKKTG
CCKGG
TGQAPGFTYTDANKNKGIIWGEDTLMEYLENPKKYIPGTKMIFAGIKKKG
CCPY
TGQAEGFSYTDANKNKGITWGEDTLMEYLENPKKYIPGTKMIFAGIKKKA
CCCH
TGQAEGFSYTDANKNKGITWGEDTLMEYLENPKKYIPGTKMIFAGIKKKS
CCDK
TGQAEGFSYTDANKNKGITWGEDTLMEYLENPKKYIPGTKMIFAGIKKKS
CCPN
TGQAEGFSYTDANKNKGITWGEDTLMEYLENPKKYIPGTKMIFAGIKKKS
CCST
TGQAEGFSYTEANKNKGITWGEETLMEYLENPKKYIPGTKMIFAGIKKKA
CCRS
TGQAVGYSYTAANKNKGIIWGDDTLMEYLENPKKYIPGTKMVFTGLKSKK
CCBN
TGQAEGYSYTDANKSKGIVWNENTLMEYLENPKKYIPGTKMIFAGIKKKG
CCFFCM
TGQAAGFAYTDANKAKGITWNEDTLFEYLENPKKYIPGTKMIFAGLKKPN
CCNC
TGSVDGYAYTDANKQKGITWDENTLFEYLENPKKYIPGTKMAFGGLKKDK
CCRZ
SGTTPGYSYSTANKNMAVIWEENTLYDYLLNPKKYIPGTKMVFPGLKKPQ
CCPO
SGTTAGYSYSNANKNMAVTWGENTLYDYLLNPKKYIPGTKMVFPGLKKPQ
CCRPBO
SGTTAGYSYSAANKNKAVEWEEKTLYDYLLNPKKYIPGTKMVFPGLKKPQ
CCWT
SGTTAGYSYSAANKNKAVEWEENTLYDYLLNPKKYIPGTKMVFPGLKKPQ
CCCRCO
SGTVEGFAYSKANADSGVVWTPEVLDVYLENPKKFMPGTKMSFAGIKKPQ
CCBY
SGQAEGYSYTDANIKKNVLWDENNMSEYLTNPKKYIPGTKMAFGGLKKEK
CCTE
AGQEK-FAYSKGMKGSGITWNEKHLFVFLKNPSKHVPGTKMAFAGLPADK
.* ..*. . . *
. .* ** * .***** * *.
CCHU
ERADLIAYLKKATNE
CCMQR
ERADLIAYLKKATNE
CCHO
EREDLIAYLKKATNE
CCHOD EREDLIAYLKKATNE
CCPG
EREDLIAYLKKATNE
CCRB
ERADLIAYLKKATNE
CCDG
ERADLIAYLKKATKE
CCKGG
ERADLIAYLKKATNE
CCPY
ERADLIAYLKQATAK
CCCH
ERVDLIAYLKDATSK
CCDK
ERADLIAYLKDATAK
CCPN ERADLIAYLKDATSK
CCST
ERADLIAYLKDATSK
CCRS
ERTDLIAYLKEATAK
CCBN
ERQDLVAYLKSATS-
CCFFCM
ERGDLIAYLKSAT-K
CCNC
DRNDIITFMKEATA-
CCRZ
ERADLISYLKEATS-
CCPO
DRADLIAYLKEATA-
CCRPBO
DRADLIAYLKEATA-
CCWT
DRADLIAYLKKATSS
CCCRCO
ERADLIAYLEN--LK
CCBY
DRNDLITYLKKACE-
CCTE
DRADLIAYLKSV---
.*
*.....
Πίνακας Δενδρογράμματος που προκύπτει από
την ευθυγράμμιση των αμινοξικών ακολουθιών των κυτοχρωμάτων C 24 οργανισμών.
91.2
0 0 2
000000000000000000120000
90.4
0 0 2
120000000000000000000000
90.4
0 0 2 001200000000000000000000
89.8
0 1 3
000000000000000001220000
89.5
0 0 2
000000000102000000000000
89.0
3 0 3
001120000000000000000000
88.8
4 0 4
000000000000000001112000
88.6
0 0 2
000000001020000000000000
88.2
8 5 4
000000001212000000000000
87.1
6 0 4
001110200000000000000000
86.8
10 0 5
001112100000000000000000
85.3
11 0 6
001111120000000000000000
84.4
9 0 5
000000001111200000000000
82.8
12 13 11 001111112222200000000000
81.5
2 14 13
112222222222200000000000
76.2
15 0 14
111111111111102000000000
75.0
16 0 15
111111111111101200000000
73.6
17 0 16
111111111111121100000000
63.7
0 0 2 000000000000000010000020
58.5
18 7 20
111111111111111102222000
57.6
20 19 22
111111111111111121111020
50.1
21 0 23
111111111111111111111210
37.5
22 0 24
111111111111111111111112
Κατασκευή φυλογενετικού δέντρου με την
μέθοδο Neighbour-Joining
DIST
= percentage divergence (/100)
Length =
number of sites used in comparison
1 vs. 2
DIST = 0.0096; length = 104
1 vs. 3
DIST = 0.1154; length = 104
1 vs. 4 DIST
= 0.1058; length = 104
1 vs. 5
DIST = 0.0962; length = 104
1 vs. 6
DIST = 0.0865; length = 104
1 vs. 7
DIST = 0.1058; length = 104
1 vs. 8
DIST = 0.0962; length = 104
1 vs. 9
DIST = 0.1154; length = 104
1 vs. 10
DIST = 0.1250; length = 104
1 vs. 11
DIST = 0.1058; length = 104
1 vs. 12
DIST = 0.1250; length = 104
1 vs. 13
DIST = 0.1442; length = 104
1 vs. 14
DIST = 0.1442; length = 104
1 vs. 15
DIST = 0.1942; length = 103
1 vs. 16
DIST = 0.2233; length = 103
1 vs. 17
DIST = 0.3689; length = 103
1 vs. 18
DIST = 0.3398; length = 103
1 vs. 19
DIST = 0.3495; length = 103
1 vs. 20
DIST = 0.3398; length = 103
1 vs. 21
DIST = 0.3365; length = 104
1 vs. 22
DIST = 0.4706; length = 102
1 vs. 23
DIST = 0.3689; length = 103
1 vs. 24
DIST = 0.5306; length = 98
2 vs. 3
DIST = 0.1058; length = 104
2 vs. 4
DIST = 0.0962; length = 104
2 vs. 5
DIST = 0.0865; length = 104
2 vs. 6
DIST = 0.0769; length = 104
2 vs. 7
DIST = 0.0962; length = 104
2 vs. 8
DIST = 0.1058; length = 104
……………
21 vs. 23
DIST = 0.3889; length = 108
21 vs. 24
DIST = 0.5943; length = 106
22 vs. 23
DIST = 0.5000; length = 106
22 vs. 24
DIST = 0.5514; length = 107
23 vs. 24
DIST = 0.6019; length = 103
Neighbor-joining
Method
Saitou, N. and
Nei, M. (1987) The Neighbor-joining Method:
A New Method
for Reconstructing Phylogenetic Trees.
Mol. Biol.
Evol., 4(4), 406-425
This is an
UNROOTED tree
Numbers in
parentheses are branch lengths
Cycle 1
= SEQ: 19 ( 0.03512) joins SEQ:
20 ( 0.03695)
Cycle 2
= SEQ: 18 ( 0.04699) joins Node: 19 (
0.00256)
Cycle 3
= Node: 18 ( 0.00607) joins SEQ: 21 ( 0.04799)
Cycle 4
= SEQ: 22 ( 0.24015) joins SEQ:
24 ( 0.31126)
Cycle 5
= SEQ: 17 ( 0.15560) joins SEQ:
23 ( 0.17151)
Cycle 6
= Node: 17 ( 0.04441) joins Node: 18 (
0.15530)
Cycle 7
= Node: 17 ( 0.00708) joins Node: 22 (
0.05344)
Cycle 8
= SEQ: 1 ( 0.00781) joins SEQ:
2 ( 0.00180)
Cycle 9
= SEQ: 3 ( 0.00837) joins SEQ:
4 ( 0.00124)
Cycle 10
= SEQ: 16 ( 0.08397) joins
Node: 17 ( 0.05686)
Cycle 11
= Node: 1 ( 0.03268) joins SEQ: 14 ( 0.11155)
Cycle 12
= SEQ: 15 ( 0.08614) joins
Node: 16 ( 0.01200)
Cycle 13
= SEQ: 10 ( 0.00910) joins SEQ:
12 ( 0.01013)
Cycle 14
= Node: 3 ( 0.01459) joins SEQ: 5 ( 0.00464)
Cycle 15
= Node: 10 ( 0.01171) joins SEQ: 11 ( 0.00753)
Cycle 16
= SEQ: 9 ( 0.01787) joins
Node: 10 ( 0.00136)
Cycle 17
= Node: 9 ( 0.01640) joins SEQ: 13 ( 0.04129)
Cycle 18
= Node: 1 ( 0.02471) joins Node: 15 (
0.02468)
Cycle 19
= Node: 1 ( 0.00439) joins Node: 9 (
0.01054)
Cycle 20
= Node: 3 ( 0.00810) joins SEQ: 7 ( 0.02075)
Cycle 21
= Node: 1 ( 0.01317) joins SEQ: 6 ( 0.01711)
Cycle 22 (Last cycle, trichotomy):
Node:
1 ( 0.00212) joins
Node:
3 ( 0.00724) joins
SEQ: 8 ( 0.03844)