

Vol.
24, No. 10, October 1999
Talking
Point
What is the true structure of
liganded haemoglobin?
Jeremy R. H. Tame
Trends in
Biochemical Sciences 1999, 24:372-377
Dept of Chemistry,
University of York, Heslington, York, UK Y010 5DD |
|


Article Outline
- Abstract
- The
R2 structure
- The
importance of the R2 structure
- Other
vertebrate haemoglobins
- Conclusion
- Acknowledgements
- References
- Copyright
Abstract
Does the crystal structure of a protein accurately represent its structure in
solution? Or does the crystallization process perturb the structure
significantly? Although aware of the problem, most crystallographers would argue
that the highly solvated and weakly held lattice in protein crystals is, in
general, unlikely to shift ordered parts of the molecule. In the case of
conformationally flexible proteins, however, there is the possibility that one
form might be favoured over another. Several lines of evidence suggest that this
might be the case for the crystal structure of liganded Hb, although conflicting
data exist.
Human haemoglobin
(Hb) is one of the most thoroughly studied biological molecules. Scientific
research into its properties dates back over a century and it even has a journal
dedicated to it. Human Hb was one of the first protein structures to be
determined by X-ray crystallography and it inspired the development of rapid
kinetics techniques and the thermodynamic theory of cooperativity. Hb is the
classic textbook example of an allosteric protein that shows the exquisite
control a protein can exert over ligand binding, although there is still
considerable debate regarding its allosteric mechanism. Biochemistry
undergraduate students are familiar with the two-state model of Monod, Wyman and
Changeux (the MWC model) in which the deoxy protein prefers a 'tense' state (T)
and binding of the ligand causes the allosteric equilibrium to move towards the
'relaxed' (R) state
1.
The higher affinity for oxygen of the R state leads to cooperative binding
according to the classical S-shaped binding curve 2.
The binding of oxygen to Hb fits the MWC model very well. When the crystal
structures of liganded and unliganded Hb were first solved and found to be
slightly different, it was natural to equate these structures with the R and T
states of the MWC model, respectively. In the liganded Hb structure, a number of
salt links between the protein subunits were broken – exactly as the MWC model
had predicted. Furthermore, the protein adopted a very similar structure in the
oxy form (which has molecular oxygen attached to Fe2+ haems and is
bright red) and in the oxidized 'met' form, which has hydrated Fe3+
haems and is brown and unable to bind oxygen. Clearly, this appeared to be the
preferred conformation of the liganded protein – the R state in MWC terminology.
In 1970, Perutz proposed to assign the crystal structures of liganded and
unliganded Hb to the R and T states 3.
Deoxy Hb in the T state switches in a concerted way to the R state as more
oxygen binds, such that the final oxygen molecules to bind Hb have the highest
affinity. Hb consists of two 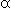
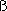 dimers that shift relative to each other to
accommodate the changes in tertiary structure of the subunits induced by either
ligand binding or ligand leaving the haem groups. The change in a key 'switch'
region, the contact between the FG corner of the subunits and the C helix of the subunits, is shown in the upper
panels of Fig.
1. Overall, the R-state tetramer, which is favoured by ligand
binding, appears somewhat looser than the T state and is more easily split into
dimers. The Perutz model has been refined by
improved deoxy- and oxy-Hb structures 3–5
and structures of intermediates on the oxygenation pathway 6–9
, as well as by functional analysis
including kinetics, NMR and a variety of spectroscopic methods. Regardless of
the pathway between the deoxy and oxy forms (and whether it involves a single
step or a sequential change, as proposed, for example, in the
Koshland–Nemethy–Filmer model), it has long been assumed that the structures of
the end-points correspond with the crystal structures, and this is the view
generally presented in textbooks. This position has been challenged over the
past few years by the discovery of a new structure of liganded Hb. Are the
textbooks wrong – is this the real structure of oxy Hb in solution?
dimers that shift relative to each other to
accommodate the changes in tertiary structure of the subunits induced by either
ligand binding or ligand leaving the haem groups. The change in a key 'switch'
region, the contact between the FG corner of the subunits and the C helix of the subunits, is shown in the upper
panels of Fig.
1. Overall, the R-state tetramer, which is favoured by ligand
binding, appears somewhat looser than the T state and is more easily split into
dimers. The Perutz model has been refined by
improved deoxy- and oxy-Hb structures 3–5
and structures of intermediates on the oxygenation pathway 6–9
, as well as by functional analysis
including kinetics, NMR and a variety of spectroscopic methods. Regardless of
the pathway between the deoxy and oxy forms (and whether it involves a single
step or a sequential change, as proposed, for example, in the
Koshland–Nemethy–Filmer model), it has long been assumed that the structures of
the end-points correspond with the crystal structures, and this is the view
generally presented in textbooks. This position has been challenged over the
past few years by the discovery of a new structure of liganded Hb. Are the
textbooks wrong – is this the real structure of oxy Hb in solution?
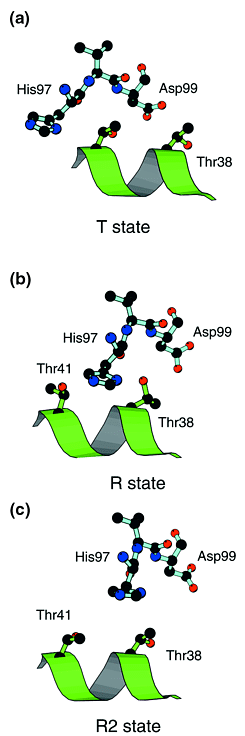 |
| Figure 1. The FG corner of the chain (shown with pale blue
bonds), which forms a key 'switch' contact with the C helix of the
neighbouring
chain (green). (a) In the T state, His97 lies close to Thr41. (b) In the R state,
the histidine has moved to the other side of Thr41 and next to Thr38. Intermediate states appear
unfavourable due to the close approach of these two residues – the
histidine must 'hop over' the threonine. (c) In the R2 state, the
histidine lies further from the C helix and so movement between the R and
T states appears to be easier. Drawn with MOLSCRIPT ( Ref.
39). |
The problem goes to the
heart of protein crystallography: how relevant are crystal structures to the
protein in solution? There has been some debate about the distortion of
structures caused by packing the molecule into a crystal lattice. A number of
proteins have been crystallized from a variety of conditions in different space
groups and show very little distortion. For example, Fedorov et al. have
shown that six crystal structures of RNase grown from different salts are
extremely similar to the form grown from alcohol 10.
Lattice forces are much more considerable for small flexible molecules. In the
case of haemoglobin, the lattice forces can actually be used to stabilize the
deoxy form of the protein, but to oxygenate the protein fully while preserving
the crystal order is a very careful balancing act that requires very slow
equilibration and low temperature 6,7
. In general, adding ligand to deoxy Hb
crystals or removing ligand from ligated ones will destroy them, unless some
means is used to stabilize them, usually a chemical cross-link of some kind. So,
a new crystal structure might well reflect the state of the protein free in
solution – lattice forces in general are too weak to force an unnatural
conformation on a protein, though they might favour one of several sampled by
the protein in solution.
The R2 structure
A new structure of Hb was discovered in
1991 by Smith, Lattman and Carter who crystallized carbonmonoxy Hb Ypsilanti (Hb
Y), a mutant of human Hb in which Asp99 is replaced by tyrosine 11.
This aspartate is highly important, forming a hydrogen bond with Tyr42 in the T state, which
stabilizes the low-affinity form of the protein. Engineered mutations of this
residue tend to lead to high affinity for oxygen and very low cooperativity,
both of which are indeed properties of Hb Y. The crystal form is clearly
distinct from the R state, having altered subunit interactions at the key
'switch' region in which His972 interacts with the C helix of
subunit 1.
In the R and T states, this histidine lies on different sides of Thr41 (C6), which appears to
form a barrier to the R T
transition ( Fig.
1). In the Hb Y structure, His972 has moved away from the C helix of
the neighbouring 1 chain, and thus Thr41 no longer presents a
steric barrier to movement of the helix along its axis. Smith and colleagues
therefore suggested 11
that the Y structure is an intermediate on the R-to-T pathway, and tentatively
correlated it with a third allosteric state suggested by the thermodynamic
analyses of Ackers and co-workers 12,13
. The severe functional disruption
caused by the mutation, however, weakens the argument that the Y form is a
physiologically important state of normal human Hb. Furthermore, the larger bulk
of a tyrosine residue at position 99 might be expected to widen the 12 interface (see Fig.
2). The new conformation is apparently stabilized by a hydrogen bond
between Tyr992 and Thr381. Much stronger evidence of a role
for this form came from Arnone et al. who crystallized carbonmonoxy
normal adult human Hb with the use of polyethylene glycol (PEG) and a relatively
low salt concentration 14.
This structure closely resembles the Y form (although the C termini of the
subunits have rather different positions) and both conformations are now
generally known as 'R2', the name given by Silva and co-workers. The overall
structures of the T, R and R2 state tetramers are compared in Fig.
3. Like Smith et al., Silva et al. suggested that this
new form of Hb might be physiologically relevant. As it could be crystallized,
this form of the protein was clearly appreciably stable in solution. Is this new
form a third allosteric state? Ackers' group has suggested that Hb occupies
three distinct energy levels on binding ligands 13.
This is disputed by Edelstein 15
and Shibayama and co-workers 16,17
but Ackers has defended his conclusions
18.
In any case, circular dichroism and sulphydryl reactivity experiments suggest
that there are significant structural perturbations of the T state of Hb Y,
which is therefore unlikely to represent the allosteric intermediate proposed by
Ackers' group 19.
T
transition ( Fig.
1). In the Hb Y structure, His972 has moved away from the C helix of
the neighbouring 1 chain, and thus Thr41 no longer presents a
steric barrier to movement of the helix along its axis. Smith and colleagues
therefore suggested 11
that the Y structure is an intermediate on the R-to-T pathway, and tentatively
correlated it with a third allosteric state suggested by the thermodynamic
analyses of Ackers and co-workers 12,13
. The severe functional disruption
caused by the mutation, however, weakens the argument that the Y form is a
physiologically important state of normal human Hb. Furthermore, the larger bulk
of a tyrosine residue at position 99 might be expected to widen the 12 interface (see Fig.
2). The new conformation is apparently stabilized by a hydrogen bond
between Tyr992 and Thr381. Much stronger evidence of a role
for this form came from Arnone et al. who crystallized carbonmonoxy
normal adult human Hb with the use of polyethylene glycol (PEG) and a relatively
low salt concentration 14.
This structure closely resembles the Y form (although the C termini of the
subunits have rather different positions) and both conformations are now
generally known as 'R2', the name given by Silva and co-workers. The overall
structures of the T, R and R2 state tetramers are compared in Fig.
3. Like Smith et al., Silva et al. suggested that this
new form of Hb might be physiologically relevant. As it could be crystallized,
this form of the protein was clearly appreciably stable in solution. Is this new
form a third allosteric state? Ackers' group has suggested that Hb occupies
three distinct energy levels on binding ligands 13.
This is disputed by Edelstein 15
and Shibayama and co-workers 16,17
but Ackers has defended his conclusions
18.
In any case, circular dichroism and sulphydryl reactivity experiments suggest
that there are significant structural perturbations of the T state of Hb Y,
which is therefore unlikely to represent the allosteric intermediate proposed by
Ackers' group 19.
.gif) |
| Figure 2. In haemoglobin Ypsilanti (Hb Y),
Asp99 is replaced
by tyrosine. In normal human deoxy haemoglobin, Asp99 forms a hydrogen bond with
Tyr42, which is
found at the end of the C helix. This hydrogen bond plays an important
role in stabilizing the T state (a), but is broken in the R state
(b). The T state has less room for a bulky side-chain at the 99 position, and is
disrupted in Hb Y (c). Drawn with MOLSCRIPT ( Ref.
39). |
.gif) |
| Figure 3. Crystal structures of human haemoglobin
with one dimer in red and the
other in blue. Orthogonal views of the tetramer are shown. The central
cavity is clearly visible in the figures on the left. (a) In
T-state haemoglobin, this cavity is larger, allowing allosteric effector
molecules such as DPG (2,3-diphosphoglycerate) to bind. (b) On
transition to the R state, or liganded state, the cavity shrinks and the
effector is expelled from its binding site. According to the Perutz
mechanism of haemoglobin allostery, the deoxy tetramer is tightly
cross-linked by a number of salt bridges. On oxygenation, movement of the
haem iron atoms relative to the rest of the haem enforce tertiary
structure changes, which break these salt bridges and cause one dimer to rotate slightly,
relative to the other. (c) Liganded subunits also permit the
tetramer to take up the R2 state conformation. Drawn with MOLSCRIPT ( Ref.
39). |
The importance of the R2 structure
The functional
importance of the R2 state is still far from established. Silva et al. 14
and Smith et al. 11
suggested that the R2 state lies between the R and T states, although
independent modelling studies by Janin and Wodak 20
and Srinivasen and Rose 21
both strongly suggested that the R2 state is a 'hyper-R' state and lies beyond
the T-to-R transition. Srinivasen and Rose concluded that the R2 state is likely
to be a better representation of the solution structure of liganded Hb than the
R state, which they suggested is an artefact of the high-salt crystallization
conditions employed by Perutz 22,
and subsequently used to solve the structure of oxy and carbonmonoxy Hb ( Refs
5,23 ). Pearson et al. 24
and Schumacher et al. 25
reached the same conclusion based on NMR experiments using
4-fluorotryptophan-labelled Hb and crystal structures of chemically cross-linked
Hb, respectively. These groups all point out that the R2 crystal form is grown
under low ('physiological') salt concentration and suggest that high salt
concentrations might impede the R-to-R2 transition.
There is, in my view,
little to support this conclusion. First, the use of 'low salt' conditions to
crystallize the protein does not necessarily make them more 'physiological'.
Indeed, significant concentrations of PEG can force oxy Hb to deoxygenate 6,26
but high salt does not do this. Colombo
and co-workers have demonstrated that neutral cosolvents such as PEG can
influence Hb very strongly 27.
Second, Hb is known to be strongly affected by pH. The R2 form of native human
Hb was grown at pH 5.8; below pH 6 the tetrameric form of the protein is
destabilized and the oxygen affinity increases. The R2 crystallization
conditions are therefore hardly physiological! The fact that Hb Y crystallizes
in the same R2 form is extremely interesting and shows that the Hb dimers can
adopt this altered packing. But, as mentioned above, Hb Y has very low
cooperativity and appears to have significant structural perturbations in the T
state 19.
This is reasonable given the known functional role played by Asp99 ( Fig.
2).
Other vertebrate haemoglobins
What, then, of the evidence
from the structures of other vertebrate Hbs? The liganded form of haemoglobins
from horse, pig, bar-headed goose and three species of fish have all been
determined by X-ray crystallography, from crystals grown under a variety of
conditions including 'low salt', in a variety of crystal symmetries 28–33
. Fish Hbs are
particularly interesting in this regard as they have diverged much further from
human Hb than Hb from air-breathing vertebrates and show a number of insertions
and deletions in both subunits. The structures of the vertebrate Hbs that have
been determined are all clearly in the R state and not R2, whether crystallized
in high salt or not ( Tables
1, 2 and 3 ). The 'switch' region contact is much closer to the R state
of human haemoglobin than R2 ( Fig.
4). These Hbs, unlike Hb Y or chemically cross-linked forms of Hb,
have all withstood the evolutionary test of time, and despite up to ~200
mutations per dimer compared with human Hb
still adopt the R state. This is much stronger evidence in support of a
physiological role for the R state than that provided for the R2 state by Hb Y.
Single mutations can often have dramatic influences on protein structure or
function, but those Hbs that Nature has selected appear to adopt the R state in
the liganded form rather than R2. The evidence provided by fluorine NMR and
chemical cross-linking that Hb adopts the R2 state in solution is also
problematic as these chemical modifications might cause distortions of the
protein or destabilize it. Pearson et al. showed that the introduction of
fluorine atoms can cause severe steric clashes, which necessitated the mutation
of Tyr130 to Phe to
eliminate an NMR peak indicative of denatured protein 24.
The fluorine atom introduced on Trp37, which was used to probe the structure, would
in fact clash with Tyr140 in the R state (but not R2) and might well
destabilize the R form in favour of R2. Neither the R nor R2 structure agreed
particularly well with the NMR data. Schumacher and co-workers determined the
X-ray structure of Hb cross-linked in the deoxy form and subsequently
crystallized the liganded form 34.
The structures have two water molecules between Asp941 and Asp992 (a hallmark of the R2 form) but,
overall, are more 'R-like' ( Table
2). A more appropriate experiment to discover the solution state of
liganded Hb is to cross-link liganded Hb rather than the deoxy form. This has
been done with horse Hb, which was crystallized following the removal of the
haem ligands. The structure turned out to be in the R state 8.
Furthermore, this deoxy R structure shows that His 97 can 'flip' out of the notch it occupies in the
R state, thereby permitting a shift towards the T structure, without passing
through the R2 form. Considerable effort has gone into crystallizing Hb
intermediates that are stabilized by lattice forces 6–8
,
cross-linking 34
or metal hybrids 35,36
. These structures offer considerable
insight into the R-to-T and T-to-R transitions but do not support a
physiological role for the R2 state.
| Table 1. Some features of crystal structures of
liganded haemoglobin a |
 |
|
|
|
|
|
|
| PDB code e |
381–972
(Å) |
Penultimate Tyr–Trp 37 (Å) b |
Resolution
(Å) |
R factor
(%) |
Salt
concentration |
pH |
|
|
|
|
|
|
|
|
| R2
structures |
|
|
|
|
|
|
|
|
| 1bbb (Hb
A) |
7.0–7.3 |
14.1–14.3 c |
1.7–6.0 |
18.4 |
Low |
5.8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1cmy (Hb
Y) |
7.0–7.2 |
6.8–7.3 |
3.0–5.0 |
26.9 |
High |
6.7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Air-breathing
vertebrates |
|
|
|
|
|
|
|
|
| 1hho (Hb
A) |
5.2 |
8.0 |
2.1–10.0 |
22.3 |
High |
6.7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1a4f
(goose) |
5.9 |
7.3 |
2.0–10.0 |
19.8 |
Low |
6.8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 2pgh
(pig) |
4.9–5.3 |
7.7–7.8 |
2.8–6.5 |
16.4 |
High |
6.8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 2mhb
(horse) |
5.4 |
8.1 |
2.0–10.0 |
23.0 |
High |
7.0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Fish d |
|
|
|
|
|
|
|
|
| 1spg
(spot) |
5.5 |
7.1 |
2.0–10.0 |
19.1 |
Low |
7.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1pbx (P.
bernacchii) |
5.3 |
7.0 |
2.5–10.0 |
17.8 |
Low |
8.0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1ouu
(trout) |
5.2–5.5 |
7.3–7.5 |
2.5–8.0 |
16.2 |
Low |
8.0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Cross-linked
human Hb A |
|
|
|
|
|
|
|
|
| 1hab |
5.8–6.4 |
7.4–7.5 |
2.3–10.0 |
19.1 |
High |
6.7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1hac |
6.1–6.8 |
7.3–7.7 |
2.6–10.0 |
15.4 |
High |
6.7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1hae |
5.7 |
7.7 |
1.8–10.0 |
17.9 |
High |
7.0–7.5 |
|
|
|
|
|
|
| |
[a]The distances shown are to
illustrate the similarity of mammalian liganded Hbs to the R form of
human Hb; the table is not intended to imply that the complexity of
information contained in a protein structure can be reduced to one or
two parameters. [b]Two distances are listed where a complete Hb
tetramer occurs in the asymmetric unit. [c]The marked movement of
Tyr140 in the
R2 structure formed by native human Hb at low salt and pH 5.8 is not
found in other liganded Hb structures. This suggests that the protein
has been destabilized by the acid conditions used and might represent an
intermediate form on the path to dissociation of the tetramer into
dimers. [d]38 and 97 are not conserved in fish Hbs, which
are more variable in polypeptide sequence and length than Hbs from
air-breathing animals. [e]Abbreviations used: Hb, haemoglobin; PDB,
Protein DataBank; P. bernacchii, Pagothenia
bernacchii.
|
| Table 2. Root-mean-square deviations of C positions (in Å) of
Hb tetramers with the canonical R and R2 structures |
|
|
|
| |
R-state tetramer a |
R2-state tetramer a |
|
|
|
|
| Animal Hbs b |
|
|
|
|
| 2pgh
(pig) |
0.89 |
2.17 |
|
|
|
|
|
|
| 2mhb
(horse) |
0.74 |
1.88 |
|
|
|
|
|
|
| 1a4f (bar-headed
goose) |
1.03 |
1.66 |
|
|
|
|
|
|
| |
|
|
|
|
|
|
| Cross-linked
human Hb |
|
|
|
|
| 1hab |
0.97 |
1.51 |
|
|
|
|
|
|
| 1hac |
1.07 |
1.50 |
|
|
|
|
|
|
| 1ibe |
0.66 |
1.81 |
|
|
| |
[a]The PDB (Protein DataBank)
accession codes for R-state oxy Hb and R2-state Hb are 1hho and 1bbb,
respectively. Both the animal Hb structures and the cross-linked human
Hb structures show higher correlations with the R state than with the R2
state. [b]The animal is shown in parentheses. The
structures described can be copied directly from PDB via the internet at
http://www.rcsb.org/pdb.
|
| Table 3. Root-mean-square deviations of C positions (in Å) of
fish Hb tetramers with the canonical R and R2 structures |
|
|
|
|
| |
No. atoms used a |
R-state tetramer
1hho |
R2-state tetramer
1bbb |
|
|
|
|
|
|
|
|
| 1pbx (P.
bernacchii) |
570 |
1.25 |
2.08 |
|
|
|
|
|
|
|
|
| 1spg
(spot) |
560 |
1.28 |
1.94 |
|
|
|
|
|
|
|
|
| 1ouu
(trout) |
574 |
1.39 |
2.26 |
|
|
|
| |
[a]The number of atoms used
varies due to insertions at various points in the fish proteins.
|
.gif) |
| Figure 4. The key switch region ( C helix – FG corner) in animal
haemoglobins. (a) Haemoglobin from the fish Leiostomus
xanthurus (a teleost fish commonly known as 'spot') in which both the
38 and 41 positions have
mutated (to glutamine and isoleucine, respectively). However, the position
of His97 relative
to these residues is similar to that found in R-state human Hb. (b)
Trout haemoglobin I (Hb I). This haemoglobin has lost many histidine
residues to minimize its Bohr effect (i.e. the pH dependence of oxygen
affinity). His97
has mutated to a phenylalanine, which in the liganded structure occupies
the same position relative to the C helix as His97 in R-state human Hb. (c)
Haemoglobin from the bar-headed goose has a high oxygen affinity due to
weakened contacts, but still
adopts the R structure in the oxy form. Drawn with MOLSCRIPT ( Ref.
39). |
Conclusion
The question 'what is the relative importance of
the R and R2 states?' remains. At present, there is a clear divergence of views.
If the R2 state is to be incorporated into the accepted view of Hb function,
then it must either displace the R state or lie to either side of it as oxy Hb
is deoxygenated. All three suggestions have been put forward:
-
TR2
-
TRR2
-
TR2R
If the R2 state
has the same oxygen affinity as the R state, then each of these schemes could
give identical oxygen-binding curves, so these are of little use in choosing the
best model. The clearest evidence for a role for the R2 state appears to be the
structure of cyanomet Hb A, reported by Smith and Simmons 37.
The protein, crystallized at pH 7.4 using PEG and 180 mm chloride, was found to adopt the R2
form, but the refinement is so far incomplete and no structure has been
deposited with the Brookhaven DataBank. The structures of animal haemoglobins,
however, seem to provide strong evidence that the R state is a better
representation of the oxy-Hb molecule. In contrast, more recent low-resolution
crystallographic studies of liganded human embryonic haemoglobin (Gower II) show
that the protein, crystallized under low-salt conditions at pH 8.5, lies between
the R and R2 structures, but closer to R2 ( Ref.
38). Until studies of Hb in solution resolve this issue, or a low-salt
structure of liganded human Hb is refined, the debate will no doubt
continue.
Acknowledgements
I would like to thank Julie Wilson, Guy Dodson, Tony Wilkinson, Maurizio
Brunori, Adriana Miele and Beatrice Vallone for helpful comments on the
manuscript, as well as the Royal Society for a University Research
Fellowship.
References
[1]
Monod J., Wyman J. and Changeux J.P. (1965)
J. Mol. Biol.,
12:88-118. [Cited
by]
[2]
Imai K. (1982) Allosteric Effects in Hemoglobin. : Cambridge University
Press
[3]
Perutz M.F. (1970)
Nature, 228:726-739. [MEDLINE] [Cited
by]
[4]
Fermi G. et al. (1984)
J. Mol. Biol.,
175:159-174. [MEDLINE] [Cited
by]
[5]
Shaanan B. (1983)
J. Mol. Biol., 171:31-59. [MEDLINE] [Cited
by]
[6]
Liddington R.C. et al. (1992)
J. Mol. Biol.,
228:551-579. [MEDLINE] [Cited
by]
[7]
Paoli M. et al. (1996)
J. Mol. Biol.,
256:775-792. [Full
text] [MEDLINE] [Cited
by]
[8]
Wilson J.C. et al. (1996)
J. Mol. Biol.,
264:743-756. [Full
text] [MEDLINE] [Cited
by]
[9]
Perutz M.F. et al. (1987)
Acc. Chem. Res.,
20:309-321. [Cited
by]
[10]
Fedorov A.A. et al. (1996)
Biochemistry,
35:15962-15979. [MEDLINE] [Cited
by]
[11]
Smith F.R., Lattman E.E. and Carter C.W. Jr (1991)
Proteins,
10:81-91. [MEDLINE] [Cited
by]
[12]
Smith F.R. and Ackers G.K. (1985)
Proc. Natl. Acad. Sci. U. S.
A., 82:5347-5351. [MEDLINE] [Cited
by]
[13]
Ackers G.K. et al. (1992)
Science, 255:54-63.
[MEDLINE] [Cited
by]
[14]
Silva M.M., Rogers P.H. and Arnone A. (1992)
J. Biol. Chem.,
267:17248-17256. [MEDLINE] [Cited
by]
[15]
Edelstein S. (1996)
J. Mol. Biol., 257:737-744. [Full
text] [MEDLINE] [Cited
by]
[16]
Shibayama N., Morimoto H. and Saigo S. (1997)
Biochemistry,
36:4375-4381. [MEDLINE] [Cited
by]
[17]
Shibayama N. et al. (1998)
Biochemistry,
37:6221-6228. [MEDLINE] [Cited
by]
[18]
Ackers G.K. (1998)
Adv. Prot. Chem., 51:185-253. [MEDLINE] [Cited
by]
[19]
Doyle M.L. et al. (1992)
Proteins, 14:351-362.
[MEDLINE] [Cited
by]
[20]
Janin J. and Wodak S.J. (1993)
Proteins, 15:1-4. [MEDLINE] [Cited
by]
[21]
Srinivasan R. and Rose G.D. (1994)
Proc. Natl. Acad. Sci. U. S.
A., 91:11113-11117. [MEDLINE] [Cited
by]
[22]
Perutz M.F. (1968)
J. Cryst. Growth, 2:54-56. [Cited
by]
[23]
Derewenda Z. et al. (1990)
J. Mol. Biol.,
211:515-519. [MEDLINE] [Cited
by]
[24]
Pearson J.G. et al. (1997)
Biochemistry,
36:3590-3599. [MEDLINE] [Cited
by]
[25]
Schumacher M.A. et al. (1997)
Proc. Natl. Acad. Sci. U. S.
A., 94:7841-7844. [Full
text] [MEDLINE] [Cited
by]
[26]
Ward K. et al. (1975)
J. Mol. Biol., 98:161-171.
[MEDLINE] [Cited
by]
[27]
Colombo M.F., Rau D.C. and Parsegian V.A. (1992)
Science,
256:655-659. [MEDLINE] [Cited
by]
[28]
Ladner R.C., Heidner E.G. and Perutz M.F. (1977)
J. Mol.
Biol., 114:385-414. [MEDLINE] [Cited
by]
[29]
Katz D.S. et al. (1994)
J. Mol. Biol.,
244:541-553. [MEDLINE] [Cited
by]
[30]
Zhang J. et al. (1996)
J. Mol. Biol.,
255:484-493. [Full
text] [MEDLINE] [Cited
by]
[31]
Mylvaganam S.E. et al. (1996)
Nat. Struct. Biol.,
3:275-283. [MEDLINE] [Cited
by]
[32]
Camardella L. et al. (1992)
J. Mol. Biol.,
224:449-460. [MEDLINE] [Cited
by]
[33]
Tame J.R.H., Wilson J.C. and Weber R.E. (1996)
J. Mol. Biol.,
259:749-760. [Full
text] [MEDLINE] [Cited
by]
[34]
Schumacher M.A. et al. (1995)
Nature, 375:85-87.
[Cited
by]
[35]
Luisi B. and Shibayama N. (1989)
J. Mol. Biol.,
206:723-736. [MEDLINE] [Cited
by]
[36]
Luisi B. et al. (1990)
J. Mol. Biol., 214:7-14.
[MEDLINE] [Cited
by]
[37]
Smith F.R. and Simmons K.C. (1994)
Proteins,
18:295-300. [MEDLINE] [Cited
by]
[38]
Sunderland-Smith A. et al. (1998)
J. Mol. Biol.,
280:475-484. [Full
text] [MEDLINE] [Cited
by]
[39]
Kraulis P.J. (1991)
J. Appl. Cryst., 24:946-950. [Cited
by]
Copyright
©
1999 Elsevier Science Ltd. All rights reserved.
